|
МЕЖГОСУДАРСТВЕННЫЙ
СОВЕТ ПО СТАНДАРТИЗАЦИИ, МЕТРОЛОГИИ И СЕРТИФИКАЦИИ |
|
|
МЕЖГОСУДАРСТВЕННЫЙ |
ГОСТ |
Определение
типов ароматических углеводородов
в средних дистиллятах.
Метод высокоэффективной жидкостной
хроматографии с обнаружением по показателю
преломления
(EN 12916:2016, IDT)
|
|
Москва Стандартинформ 2018 |
Цели, основные принципы и основной порядок проведения работ по межгосударственной стандартизации установлены в ГОСТ 1.0-2015 «Межгосударственная система стандартизации. Основные положения» и ГОСТ 1.2-2015 «Межгосударственная система стандартизации. Стандарты межгосударственные, правила и рекомендации по межгосударственной стандартизации. Правила разработки, принятия, обновления и отмены»
1 ПОДГОТОВЛЕН Научно-производственным республиканским унитарным предприятием «Белорусский государственный институт стандартизации и сертификации» (БелГИСС) на основе собственного перевода на русский язык англоязычной версии стандарта, указанного в пункте 5
2 ВНЕСЕН Государственным комитетом по стандартизации Республики Беларусь
3 ПРИНЯТ Межгосударственным советом по стандартизации, метрологии и сертификации (протокол от 30 марта 2017 г. № 97-П)
За принятие проголосовали:
|
Краткое наименование страны |
Код страны |
Сокращенное наименование национального органа |
|
Армения |
AM |
Минэкономики Республики Армения |
|
Беларусь |
BY |
Госстандарт Республики Беларусь |
|
Казахстан |
KZ |
Госстандарт Республики Казахстан |
|
Киргизия |
KG |
Кыргызстандарт |
|
Россия |
RU |
Росстандарт |
|
Таджикистан |
TJ |
Таджикстандарт |
|
Узбекистан |
UZ |
Узстандарт |
(Поправка).
4 Приказом Федерального агентства по техническому регулированию и метрологии от 31 октября 2018 г. № 902-ст межгосударственный стандарт ГОСТ EN 12916-2017 введен в действие в качестве национального стандарта Российской Федерации с 1 января 2019 г.
5 Настоящий стандарт идентичен европейскому стандарту EN 12916:2016 «Нефтепродукты. Определение типов ароматических углеводородов в средних дистиллятах. Метод высокоэффективной жидкостной хроматографии с обнаружением по показателю преломления» («Petroleum products - Determination of aromatic hydrocarbon types in middle distillates - High performance liquid chromatography method with refractive index detection», IDT).
Европейский стандарт разработан Техническим комитетом по стандартизации CEN/TC 19 «Газообразные и жидкие топлива, смазочные материалы и относящиеся к ним нефтепродукты синтетического и биологического происхождения» Европейского комитета по стандартизации (CEN).
При применении настоящего стандарта рекомендуется использовать вместо ссылочных европейских стандартов соответствующие им межгосударственные стандарты, сведения о которых приведены в дополнительном приложении ДА
6 ВВЕДЕН ВПЕРВЫЕ
Информация об изменениях к настоящему стандарту публикуется в ежегодном информационном указателе «Национальные стандарты», а текст изменений и поправок - в ежемесячном информационном указателе «Национальные стандарты». В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ежемесячном информационном указателе «Национальные стандарты». Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет (www.gost.ru)
ГОСТ EN 12916-2017
Определение типов ароматических углеводородов в средних
дистиллятах.
Метод высокоэффективной жидкостной хроматографии с обнаружением
по показателю преломления
Petroleum products. Determination of aromatic hydrocarbon types in middle distillates.
High performance liquid chromatography method with refractive index detection
Дата введения - 2019-01-01
1 Область применения
Настоящий стандарт устанавливает метод определения содержания моноароматических, диароматических, три+-ароматических углеводородов в дизельных топливах, в том числе содержащих до 30 % (V/V) метиловых эфиров жирных кислот (FAME), и нефтяных дистиллятах с диапазоном кипения от 150 °С до 400 °С. Содержание полициклических ароматических углеводородов вычисляют суммированием содержания диароматических и три+-ароматических углеводородов, а общее содержание ароматических соединений - суммированием содержания индивидуальных групп ароматических углеводородов.
Серосодержащие, азотсодержащие и кислородсодержащие соединения могут мешать определению; моноалкены не мешают определению, однако диалкены или полиалкены с сопряженными двойными связями (в случае их присутствия) могут влиять на результат определения.
Показатели прецизионности метода испытания настоящего стандарта были установлены для дизельных топлив, как содержащих, так и не содержащих FAME в качестве смесевого компонента, с содержанием моноароматических соединений в диапазоне значений от 6 % (m/m) до 30 % (m/m), диароматических соединений в диапазоне значений от 1 % (m/m) до 10 % (m/m), три+-ароматических соединений в диапазоне значений от 0 % (m/m) до 2 % (m/m), полициклических ароматических соединений в диапазоне значений от 1 % (m/m) до 12 % (m/m) и с общим содержанием ароматических соединений в диапазоне значений от 7 % (m/m) до 42 % (m/m).
Примечания
1 В настоящем стандарте единицы измерения % (m/m) и % (V/V) применяют для обозначения массовой и объемной доли соответственно.
2 Группы ароматических соединений принято определять на основе их элюционных характеристик при хроматографировании на заданной колонке для жидкостной хроматографии в сравнении с соответствующими характеристиками эталонных ароматических соединений. Количественное определение групп ароматических соединений проводится методом внешней калибровки с использованием одного ароматического соединения для каждой группы, которое не обязательно является представителем ароматических соединений, присутствующих в пробе. При использовании других аппаратуры и методик испытания разделение по группам и количественное определение индивидуальных групп ароматических соединений может быть иным.
Предупреждение - При применении настоящего стандарта могут использоваться опасные вещества, операции и оборудование. Настоящий стандарт не предусматривает рассмотрение всех проблем безопасности, связанных с его применением. Ответственность за установление мер по обеспечению техники безопасности и охраны здоровья, а также определение ограничений по применению стандарта несет пользователь настоящего стандарта.
2 Нормативные ссылки
В настоящем стандарте использованы нормативные ссылки на следующие стандарты:
EN 14214, Liquid petroleum products - Fatty acid methyl esters (FAME) for use in diesel engines and heating applications - Requirements and test methods (Топлива для двигателей внутреннего сгорания. Метиловые эфиры жирных кислот (FAME) для дизельных двигателей. Технические требования и методы испытаний)
EN ISO 1042, Laboratory glassware - One-mark volumetric flasks (Посуда лабораторная стеклянная. Колбы мерные с одной меткой) (ISO 1042)
EN ISO 3170, Petroleum liquids - Manual sampling (Нефтепродукты жидкие. Ручной отбор проб) (ISO 3170)
EN ISO 3171, Petroleum liquids -Automatic pipeline sampling (Нефтепродукты жидкие. Автоматический отбор проб из трубопроводов) (ISO 3171)
3 Термины и определения
В настоящем стандарте применены следующие термины с соответствующими определениями:
3.1 неароматические углеводороды (non-aromatic hydrocarbon): Соединения, время удерживания которых на заданной полярной колонке меньше времени удерживания большинства моноароматических соединений.
3.2 моноароматические углеводороды; MAH (mono-aromatic hydrocarbon): Соединения, время удерживания которых на заданной полярной колонке больше времени удерживания большинства неароматических соединений, но меньше времени удерживания большинства диароматических соединений.
3.3 диароматические углеводороды; DAH (di-aromatic hydrocarbon): Соединения, время удерживания которых на заданной полярной колонке больше времени удерживания большинства моноароматических соединений, но меньше времени удерживания большинства три+-ароматических соединений.
3.4 три+-ароматические углеводороды; T+AH (tri+aromatic hydrocarbon): Соединения, время удерживания которых на заданной полярной колонке больше времени удерживания большинства диароматических соединений, но меньше времени удерживания хризена.
3.5 полициклические ароматические углеводороды; POLY-АН (polycyclic aromatic hydrocarbon): Сумма диароматических и три+-ароматических углеводородов.
3.6 общее содержание ароматических углеводородов (total aromatic hydrocarbon): Сумма моноароматических, диароматических и три+-ароматических углеводородов.
Примечание - Различные опубликованные и неопубликованные данные указывают на то, что к основным компонентам каждой группы углеводородов могут относиться:
а) к неароматическим углеводородам - ациклические и циклические алканы (парафины и нафтены), моноалкены (при их наличии);
b) к MAH - бензолы, тетралины, инданы и высшие нафтенбензолы (например, октагидрофенантрен), тиофены, стиролы и полиалкены с сопряженными двойными связями;
с) к DAH - нафталины, бифенилы, индены, флуорены, аценафтены, бензотиофены и дибензотиофены;
d) к T+AH - фенантрены, пирены, флуорантены, хризены, трифенилены, бензантрацены.
3.7 метиловые эфиры жирных кислот; FAME (fatty acid methyl esters): Смесь метиловых эфиров жирных кислот, полученных из растительных масел, удовлетворяющая требованиям EN 14214.
4 Сущность метода
Навеску пробы известной массы разбавляют гептаном и заданный объем полученного раствора вводят в высокоэффективный жидкостный хроматограф, оснащенный полярной колонкой. Данная колонка обладает низким сродством к неароматическим углеводородам, но проявляет высокую селективность по отношению к ароматическим углеводородам. Вследствие указанной селективности ароматические углеводороды отделяются от неароматических углеводородов и разделяются на отдельные зоны в зависимости от числа ароматических колец, т. е. на зоны, соответствующие соединениям MAH, DAH и T+AH.
Колонка соединена с рефрактометрическим детектором, который детектирует компоненты по мере их элюирования из колонки. Сигнал детектора непрерывно регистрируется системой накопления данных. Амплитуды сигналов, соответствующих ароматическим соединениям в пробе, сравниваются с амплитудами сигналов, полученными при испытании калибровочных стандартных растворов, с целью дальнейшего вычисления массовых долей MAH, DAH и T+AH в пробе. Сумму массовых долей DAH и T+AH указывают в протоколе испытания как массовую долю POLY-АН, а сумму массовых долей MAH, DAH и T+AH указывают в протоколе испытания как массовую долю всех ароматических углеводородов.
После того как ароматические соединения элюируются из колонки, ее можно промыть обратным потоком, чтобы элюировались оставшиеся соединения, такие как FAME, в виде пика обратного потока. Данная процедура позволяет лучше очистить колонку, но следует соблюдать осторожность, так как она может повлиять на срок службы колонки.
5 Реактивы и материалы
Необходимо использовать доступные реактивы и материалы с наивысшей степенью чистоты; реактивы для метода высокоэффективной жидкостной хроматографии (HPLC) могут быть приобретены у крупных поставщиков.
5.2 Циклогексан со степенью чистоты не менее 99 % (m/m) (CAS № 110-82-7).
Примечание - Циклогексан может содержать в качестве примеси бензол.
5.3 Гептан для высокоэффективной жидкостной хроматографии (HPLC), используемый в качестве подвижной фазы (CAS № 142-82-5).
Изменение качества растворителя от партии к партии в отношении содержания воды, вязкости, показателя преломления и степени чистоты может привести к непредсказуемому поведению колонки. Сушка (например, выдерживанием над активированным молекулярным ситом типа 5А) и фильтрование мобильной фазы могут уменьшить влияние следовых количеств примесей, присутствующих в растворителе.
Подвижную фазу перед использованием рекомендуется дегазировать; данная процедура может быть проведена встроенной в прибор системой или отдельно путем пропускания (барботирования) через подвижную фазу гелия, методом вакуумной дегазации или посредством обработки ультразвуком. Несоблюдение рекомендаций по дегазации может привести к появлению отрицательных пиков.
5.4 1-Фенилдодекан со степенью чистоты не менее 98 % (m/m) (CAS № 123-01-3).
5.5 1,2-Диметилбензол (о-ксилол) со степенью чистоты не менее 98 % (m/m) (CAS № 95-47-6).
5.6 Гексаметилбензол со степенью чистоты не менее 98 % (m/m) (CAS № 87-85-4).
5.7 Нафталин со степенью чистоты не менее 98 % (m/m) (CAS № 91-20-3).
5.8 Флуорен со степенью чистоты не менее 98 % (m/m) (CAS № 86-73-7).
5.9 Фенантрен со степенью чистоты не менее 98 % (m/m) (CAS № 85-01-8).
5.10 Дибензотиофен со степенью чистоты не менее 95 % (m/m) (CAS № 132-65-0).
5.11 9-Метилантрацен со степенью чистоты не менее 95 % (m/m) (CAS № 779-02-2).
5.12 Хризен со степенью чистоты не менее 95 % (m/m) (CAS № 218-01-9).
5.13 FAME, удовлетворяющие требованиям EN 14214.
Предупреждение - При работе с ароматическими соединениями следует пользоваться защитными перчатками.
6 Аппаратура
6.1 Жидкостный хроматограф, укомплектованный высокоэффективной системой, обеспечивающей прокачивание подвижной фазы со скоростью от 0,5 до 1,5 см3/мин, с точностью выше 0,5 %, и пульсацией менее 1 % от верхнего предела шкалы в условиях испытаний, приведенных в разделе 8.
6.2 Система ввода пробы, обеспечивающая впрыскивание раствора пробы объемом 10 мм3 со значением повторяемости менее 1 %.
В хроматограф должны вводиться равные и постоянные объемы калибровочного раствора и раствора пробы. Системы с ручным и автоматическим вводом пробы (с полным или частичным заполнением петли для пробы) могут удовлетворять требованиям указанной повторяемости, если они используются правильно. При использовании способа с частичным заполнением петли рекомендуется, чтобы объем вводимой пробы составлял менее половины общего объема петли. При полном заполнении петли лучшие результаты достигаются при введении пробы объемом, превышающим объем петли не менее чем в шесть раз.
Повторяемость системы ввода пробы может быть проверена сравнением площадей пиков, полученных после ввода стандартного раствора для калибровки хроматографической системы не менее четырех раз (см. 8.3).
Допускается использовать пробы и калибровочные растворы объемом, отличающимся от 10 мм3 (как правило, в диапазоне от 3 до 20 мм3), при условии соответствия требованиям к повторяемости ввода, чувствительности и линейности в отношении показателя преломления (см. 9.4) и разрешающей способности колонки (см. 8.9).
6.3 Фильтр для пробы, используемый при необходимости (см. 10.1), представляющий собой микрофильтр с размером пор 0,45 мкм или менее, химически инертный к углеводородным растворителям, предназначенный для удаления механических примесей из растворов пробы.
Примечание - Установлено, что политетрафторэтиленовые фильтры являются пригодными для проведения испытания.
6.4 Система колонок, состоящая из колонки (колонок) для высокоэффективной жидкостной хроматографии из нержавеющей стали, заполненной неподвижной кремниевой фазой с привитыми аминогруппами (или амино/цианогруппами) и размерами гранул 3, 5 или 10 мкм, удовлетворяющая требованиям к разрешающей способности, приведенным в 8.6, 8.7, 8.9 и 8.11. Указания по выбору и использованию соответствующей системы колонок приведены в приложении А.
6.5 Устройство регулирования температуры для различных частей аппаратуры (колонка, устройство ввода пробы, растворителя, рефрактометрический детектор). Температуру устройства ввода пробы поддерживают такой же, как у раствора пробы, для колонки может использоваться нагревательный блок или термостат с циркулирующим воздухом. Также может использоваться лабораторная установка искусственного климата, обеспечивающая поддержание постоянной температуры в диапазоне от (20 ± 1) °С до (40 ± 1) °С.
Рефрактометрический детектор чувствителен как к резким, так и к плавным изменениям температуры элюента. Поэтому следует предпринимать все необходимые меры для поддержания постоянного температурного режима в пределах всей хроматографической системы. Оптимальное значение температуры должно подбираться в зависимости от неподвижной фазы.
6.6 Рефрактометрический детектор, обеспечивающий регистрацию значений показателя преломления в диапазоне от 1,3 до 1,6, обладающий линейным откликом в пределах калибровочного диапазона и выходным сигналом, приемлемым для накопления данных.
Примечание - Если рефрактометрический детектор оснащен независимой системой регулирования температуры, рекомендуется устанавливать для данной системы такое же значение температуры, как и для термостата колонки.
6.7 Компьютер или вычислительный интегратор, совместимый с рефрактометрическим детектором, обеспечивающий регистрацию данных с частотой 1 Гц и измерение площади пиков и времени удерживания. Компьютер или интегратор должен обеспечивать возможность обработки данных после анализа (например, коррекцию базовых линий и проведение повторного интегрирования).
Примечание - Наличие функции автоматического детектирования и идентификации пиков, а также вычисления концентраций соединений, исходя из площадей пиков, является рекомендуемым, но не обязательным.
6.8 Мерные колбы вместимостью 10 и 100 см3, класса А, по EN ISO 1042.
6.9 Аналитические весы с погрешностью ±0,0001 г.
7 Отбор проб
При отсутствии других указаний в технических условиях на товарный продукт отбор проб должен осуществляться в соответствии с процедурами, приведенными в EN ISO 3170 или EN ISO 3171, и/или требованиями национальных стандартов, или правилами отбора проб испытуемого продукта.
8 Подготовка аппаратуры
8.1 Устанавливают и настраивают жидкостный хроматограф (см. 6.1), систему ввода пробы (см. 6.2), колонку (см. 6.4), рефрактометрический детектор (см. 6.6) и вычислительный интегратор (см. 6.7) в соответствии с инструкциями изготовителя оборудования. При использовании термостата для колонки (см. 6.5) ее устанавливают в данный термостат. Систему ввода пробы поддерживают при температуре, равной температуре раствора пробы; в большинстве случаев данная температура будет совпадать с комнатной.
Примечание - Для обеспечения надлежащего функционирования жидкостного хроматографа и его компонентов необходимо регулярно осуществлять их техническое обслуживание. Утечки и частичное засорение фильтров, фритты, инжекторных игл и вентилей могут привести к изменению скорости потока и ухудшению повторяемости.
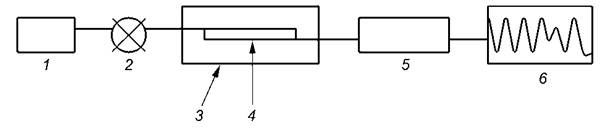
1 - насос; 2 - устройство ввода; 3 - термостат; 4 - колонка; 5 - рефрактометрический детектор; 6 - система сбора данных
Рисунок 1 - Схематическое изображение жидкостного хроматографа
8.2 Устанавливают постоянное значение скорости потока подвижной фазы в диапазоне от 0,8 до 1,2 см3/мин и убеждаются в том, что кювета сравнения рефрактометрического детектора полностью заполнена подвижной фазой. Ожидают, пока температура колонки и рефрактометрического детектора (в том случае, если детектор оснащен устройством регулирования температуры) не стабилизируется.
Для сведения к минимуму дрейфа показаний прибора кювета сравнения детектора должна быть заполнена подвижной фазой, что достигается либо путем пропускания подвижной фазы через кювету сравнения непосредственно перед анализом, с последующей изоляцией кюветы для предотвращения испарения жидкости, либо путем непрерывного пропускания потока подвижной фазы через кювету сравнения для компенсации потерь вследствие испарения. Оптимальное значение скорости потока подбирают таким образом, чтобы ошибки в результате высыхания кюветы (сравнения), а также колебания температуры или перепады давления (в кювете сравнения или измерительной кювете - в зависимости от типа детектора) были минимальными; для некоторых детекторов выполнение данных условий может быть достигнуто при скорости потока подвижной фазы, проходящей через кювету сравнения, равной одной десятой от скорости потока через измерительную кювету.
8.3 Для приготовления стандартного раствора 1 для калибровки хроматографической системы (SCS1) в мерной колбе вместимостью 100 см3 взвешивают с округлением результата до 0,001 г:
- (1,0 ± 0,1) г циклогексана (см. 5.2);
- (0,1 ± 0,01) г 1-фенилдодекана (см. 5.4);
- (0,5 ± 0,05) г 1,2-диметилбензола (см. 5.5);
- (0,1 ± 0,01) г гексаметилбензола (см. 5.6);
- (0,1 ± 0,01) г нафталина (см. 5.7);
- (0,05 ±0,005) г дибензотиофена (см. 5.10);
- (0,05 ± 0,005) г 9-метилантрацена (см. 5.11).
Помещают колбу с ее содержимым в ультразвуковую баню и выдерживают до тех пор, пока при визуальном осмотре не станет видно, что все компоненты растворились в смеси 1,2-диметилбензола и циклогексана. Извлекают колбу из ультразвуковой бани и доводят гептаном ее содержимое до метки.
SCS1 может храниться не менее 1 года в плотно укупоренной бутылке в прохладном темном месте (например, в холодильнике).
8.4 Для приготовления стандартного раствора 2 для калибровки хроматографической системы (SCS2) в мерной колбе вместимостью 100 см3 взвешивают с округлением результата до 0,001 г (0,4 ± 0,1) г FAME (см. 5.13), (0,04 ± 0,01) г хризена (см. 5.12) и доводят содержимое до метки гептаном (см. 5.3). Выдерживают раствор в ультразвуковой бане при температуре 35 °С.
Удостоверяются при визуальном осмотре, что раствор является однородным и не содержит отложений хризена на дне.
Примечание - Установлено, что 25 мин является достаточным временем для растворения всех компонентов.
SCS2 может храниться не менее 1 года в плотно укупоренной бутылке в прохладном темном месте (например, в холодильнике).
8.5 После того как рабочие параметры стабилизируются, о чем свидетельствует появление стабильной горизонтальной базовой линии, вводят 10 мм3 SCS1 (см. 8.3). Следят за тем, чтобы дрейф базовой линии в течение анализа составлял не более 1 % от высоты пика циклогексана.
Примечание - Дрейф базовой линии, превышающий 1 %, указывает на наличие неполадок в системе регулирования температуры колонки/рефрактометрического детектора и/или на элюирование какого-либо соединения из колонки.
8.6 Убеждаются в том, что все компоненты SCS1 элюируются в следующей последовательности: циклогексан, фенилдодекан, 1,2-диметилбензол, гексаметилбензол, нафталин, дибензотиофен и 9-метилантрацен.
8.7 Убеждаются в том, что разделение всех компонентов SCS1 происходит до базовой линии (см. рисунок 2).
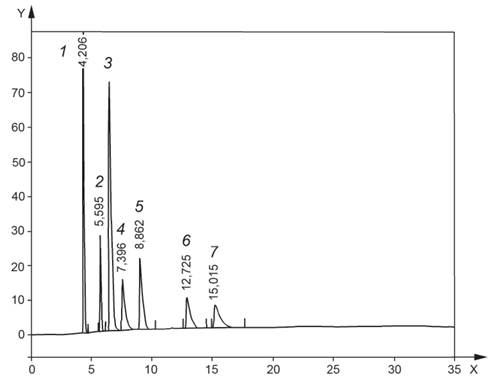
1 -
циклогексан; 2 - фенилдодекан; 3 - 1,2-диметилбензол; 4 -
гексаметилбензол; 5 - нафталин; 6 - дибензотиофен;
7 - 9-метилантрацен; ось X - время, мин; ось Y - сигнал, мВ
Рисунок 2 - Хроматограмма калибровочного стандартного раствора SCS1
8.8 Измеряют значения времени удерживания пиков циклогексана, фенилдодекана, 1,2-диметилбензола, гексаметилбензола, дибензотиофена и 9-метилантрацена с использованием системы обработки данных.
8.9 Убеждаются в том, что степень разделения (разрешающая способность) циклогексана и 1,2-диметилбензола составляет от 5,7 до 10 (см. 11.2).
8.10 Вычисляют отрезки времени, используя формулы, приведенные в 11.3.
8.11 Убеждаются в том, что раствор SCS2 визуально является однородным (см. 8.4), после чего вводят 10 мм3 SCS2 и проверяют, каким образом элюируется пик хризена - непосредственно перед первым пиком FAME или вместе с ним.
Обеспечивают, чтобы время удерживания пика хризена было больше времени удерживания пика 9-метилантрацена.
Перед проведением испытаний на новой колонке после периода простоя или перед испытанием проб, содержащих FAME, проверяют работоспособность колонки путем испытания SCS2.
9 Калибровка
9.1 Готовят четыре калибровочных стандартных раствора (A, B, C и D) с точно известными концентрациями, приблизительные значения которых приведены в таблице 1, взвешиванием с точностью до 0,0001 г в мерных колбах вместимостью 100 см3 соответствующих соединений и доведением объема растворов до метки гептаном (см. 5.3).
Примечание - Калибровочные стандартные растворы сохраняют свои свойства в течение не менее 6 мес при хранении в плотно укупоренных сосудах (например, в мерных колбах вместимостью 100 см3) в прохладном темном месте (например, в холодильнике).
9.2 После того как рабочие параметры стабилизируются (см. 8.5), вводят 10 мм3 калибровочного стандартного раствора А. Записывают хроматограмму и измеряют площадь пика для каждого ароматического соединения в калибровочном стандартном растворе (см. рисунок 3).
Таблица 1 - Концентрации компонентов в калибровочных стандартных растворах
|
Калибровочный |
1,2-Диметилбензол, г/100 см3 |
Флуорен, г/100 см3 |
Фенантрен, г/100 см3 |
|
А |
4,0 |
2 |
0,4 |
|
В |
1,0 |
1,0 |
0,2 |
|
С |
0,25 |
0,25 |
0,05 |
|
D |
0,05 |
0,02 |
0,01 |
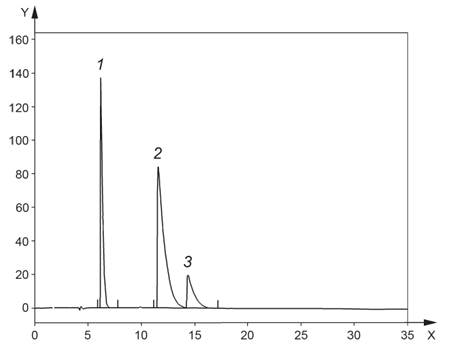
1 - 1,2-диметилбензол; 2 - флуорен; 3 - фенантрен; ось Х - время, мин; ось У - сигнал, мВ
Рисунок 3 - Хроматограмма калибровочного стандартного раствора
9.3 Повторяют процедуру, описанную в 9.2, для всех других калибровочных стандартных растворов - В, С и D.
Если площадь пика фенантрена в калибровочном стандартном растворе D слишком мала для ее точного измерения, готовят новый калибровочный стандартный раствор D+ с более высокой концентрацией фенантрена, например 0,02 г/100 см3, и повторяют процедуру, описанную в 9.2.
9.4 Строят график зависимости рассчитанной площади пика от концентрации в г/100 см3 для каждого ароматического соединения в растворе, т. е. для 1,2-диметилбензола, флуорена и фенантрена.
Калибровочные кривые должны быть линейными с коэффициентом корреляции выше 0,999 и отклонение точки их пересечения с осью ординат от начала координат должно быть в интервале ±0,01 г/100 см3. Для построения калибровочных кривых может использоваться компьютер или система обработки данных.
10 Проведение испытания
10.1 Взвешивают от 0,9 до 1,1 г пробы с точностью до 0,001 г в мерной колбе вместимостью 10 см3 и доводят содержимое гептаном (см. 5.3) до метки. Энергично встряхивают колбу для перемешивания ее содержимого. Оставляют раствор на 10 мин и при необходимости с использованием фильтра (см. 6.3) удаляют нерастворимые вещества.
Если концентрация одной или нескольких групп ароматических углеводородов выходит за пределы калибровочного диапазона, готовят в зависимости от конкретной ситуации более концентрированный (2 г/10 см3) или более разбавленный (0,5 г/10 см3) раствор пробы.
Примечание - Использование коэффициента разбавления, отличного от предложенного, может привести к изменению времени удерживания и результата вычисления содержания углеводородов.
10.2 После того как рабочие параметры стабилизируются (см. 8.5) и станут идентичными тем параметрам, которые использовались для получения калибровочных данных (см. раздел 9), вводят 10 мм3 раствора пробы (см. 10.1) и начинают сбор данных.
10.3 Проводят надлежащим образом идентификацию групп MAH, DAH и T+AH:
- MAH - соединения, время удерживания которых находится в интервале от tb до tc (см. 11.3);
- DAH - соединения, время удерживания которых находится в интервале от tc до td (см. 11.3);
- T+AH - соединения, имеющие время удерживания между td и te (см. 11.3).
10.4 После того как все ароматические соединения элюируются из колонки, она может быть промыта обратным потоком, чтобы элюировались оставшиеся соединения, такие как FAME, в виде пика обратного потока.
Промывка обратным потоком позволяет лучше очистить колонку, но следует соблюдать осторожность, так как это может повлиять на срок службы колонки.
10.5 Проводят линию от точки, расположенной непосредственно перед началом пика неароматических углеводородов (точка A (ta) на рисунке 4), до точки на хроматограмме, расположенной сразу же после T+AH (точка E (te) на рисунке 4), в которой сигнал возвращается к базовой линии (т. е. сигнал в точке A допускается для любого дрейфа базовой линии (см. 8.5)).
Если DAH и/или T+AH в пробе отсутствуют, то точка E должна быть выбрана при более раннем времени удерживания, при котором значение сигнала возвращается к базовой линии (т. е. сигнал в точке A допускается для любого дрейфа базовой линии (см. 8.5)).
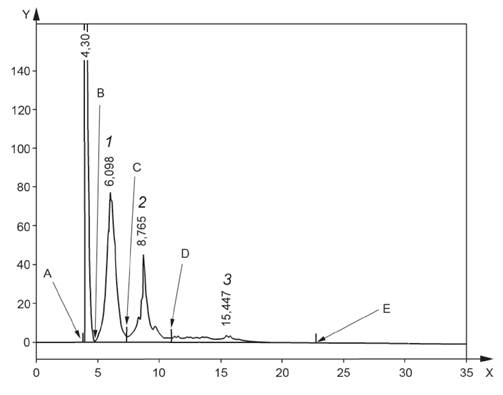
1 -
моноароматические углеводороды; 2 - диароматические углеводороды; 3
- три+-ароматические углеводороды;
ось X - время, мин; ось Y - сигнал, мВ; A - начало пика
неароматических углеводородов, ta, B - начало пика MAH, tb;
C - впадина между MAH и DAH tc, D - впадина между DAH и T+AH td; Е - конец площади интегрирования te
Рисунок 4 - Хроматограмма с идентифицированными пиками и отображением интегрирования
10.6 Проводят вертикальную линию от впадины между пиком неароматических углеводородов и пиком MAH (точка В (tb) на рисунке 4) до базовой линии (см. 10.5). При наличии нескольких впадин выбирают ту впадину, которая находится ближе всех ко времени tb (см. 11.3).
10.7 Проводят вертикальную линию от впадины между пиком MAH и пиком DAH (точка С (tc) на рисунке 4) до базовой линии (см. 10.5). При наличии нескольких впадин выбирают ту впадину, которая находится ближе всех ко времени tc (см. 11.3).
10.8 Проводят вертикальную линию от впадины между пиком DAH и пиком T+AH (точка D (td) на рисунке 4) до базовой линии (см. 10.5). При наличии нескольких впадин выбирают ту впадину, которая находится ближе всех ко времени td (см. 11.3). При отсутствии впадин выбирают время td.
10.9 Интегрируют площадь, соответствующую MAH, от точки В до точки С.
10.10 Интегрируют площадь, соответствующую DAH, отточки С до точки D.
10.11 Интегрируют площадь, соответствующую T+AH, от точки D до точки Е.
10.12 Если обработка хроматографических данных была выполнена автоматически, проверяют визуально правильность идентификации и интегрирования пиков.
11 Вычисления
Значения времени удерживания, измеренные по хроматограмме SCS1 (см. 8.8), следующие:
- время удерживания циклогексана t1, с;
- время удерживания фенилдодекана t2, с;
- время удерживания 1,2-диметилбензола t3, с;
- время удерживания гексаметилбензола t4, с;
- время удерживания дибензотиофена t6, с;
- время удерживания 9-метилантрацена t7, с.
11.2 Разрешающая способность колонки
Вычисляют разрешающую способность R между циклогексаном и 1,2-диметилбензолом по следующей формуле:
|
|
(1) |
где y1 - ширина пика циклогексана на половине высоты, с;
y2 - ширина пика 1,2-диметилбензола на половине высоты, с.
Определяют отрезки времени ta, tb, tc, td и te, с, следующим образом:
- ta - точка на базовой линии перед пиком неароматических углеводородов;
- tb = 0,5 (t1 + t2);
- tc = t4
- td = t6 + 0,4(t7 - t6),
- te - точка на базовой линии после элюирования всех T+AH.
Примечание - Для проб, содержащих FAME, te - точка на базовой линии перед первым пиком FAME.
11.4 Содержание отдельных групп ароматических углеводородов
Определяют массовую долю С MAH, DAH и T+AH, используя систему обработки данных, или вычисляют по следующей формуле:
|
|
(2) |
где A - площадь пика MAH, DAH или Т+АН, содержащихся в пробе;
S - угол наклона калибровочной кривой для MAH, DAH или Т+АН (кривой зависимости площади пика от концентрации в г/100 см3);
l - отрезок, отсекаемый калибровочной кривой для MAH, DAH и Т+АН на оси ординат;
М - масса отобранной пробы, г (см. 10.1);
V - объем раствора пробы, см3 (см. 10.1).
11.5 Содержание полициклических ароматических углеводородов и общее содержание ароматических углеводородов
Вычисляют содержание полициклических ароматических углеводородов в пробе как массовую долю путем суммирования содержания диароматических и три+-ароматических углеводородов (т. е DAH и Т+АН), а также общее содержание ароматических углеводородов в пробе как массовую долю путем суммирования массовых долей индивидуальных групп ароматических углеводородов (т. е. МАН, DAH и Т+АН).
12 Выражение результатов
Записывают содержание MAH, DAH, Т+АН, POLY-АН и общее содержание ароматических углеводородов с точностью до 0,1 % (m/m).
13 Прецизионность
Показатели прецизионности, полученные в результате статистической обработки результатов межлабораторных испытаний в соответствии с [2], приведены в 13.2 и 13.3.
Расхождение между двумя результатами испытания, полученными одним и тем же оператором при работе на одном и том же оборудовании при одинаковых условиях испытания на идентичном испытуемом продукте при нормальном и правильном выполнении метода в течение длительного времени, только в одном случае из двадцати может превысить значения, приведенные в таблице 2.
Расхождение между двумя отдельными и независимыми результатами испытаний, полученными разными операторами в разных лабораториях на идентичном испытуемом продукте при нормальном и правильном выполнении метода в течение длительного времени, только в одном случае из двадцати может превысить значения, приведенные в таблице 2.
Таблица 2 - Показатели прецизионности
|
Группа ароматических углеводородов |
Диапазон, % (m/m) |
Повторяемость |
Воспроизводимость |
|
Моноароматические углеводороды (MAH) |
От 6 до 30 |
0,032X - 0,161 |
0,144X - 0,344 |
|
Диароматические углеводороды (DAH) |
От 1 до 10 |
0,151X - 0,036 |
0,363X - 0,087 |
|
Три+-ароматические углеводороды (T+AH) |
От 0 до 2 |
0,092X + 0,098 |
0,442X + 0,471 |
|
Полициклические ароматические углеводороды (POLY-AH) |
От 1 до 12 |
0,074X + 0,186 |
0,185X + 0,465 |
|
Общее содержание ароматических углеводородов |
От 7 до 42 |
0,040X - 0,070 |
0,172X - 1,094 |
|
Примечание - X - среднее значение двух сравниваемых результатов. |
|||
14 Протокол испытаний
Протокол испытаний должен содержать по меньшей мере следующую информацию:
a) тип и полную идентификацию испытуемого продукта;
b) ссылку на настоящий стандарт;
c) результат испытания (см. раздел 12);
d) любые отклонения от установленного метода;
e) дату испытания.
Приложение A
(справочное)
Выбор и использование колонки
Колонки длиной от 150 до 300 мм и внутренним диаметром от 4 до 5 мм показали удовлетворительные результаты. Для защиты аналитической колонки рекомендуется применять защитную колонку (например, длиной 30 мм и внутренним диаметром 4,6 мм, наполненную оксидом кремния с привитой аминогруппой), которую регулярно меняют.
Для имеющихся в продаже марок неподвижной фазы были отмечены колебания от партии к партии разрешающей способности и селективности в отношении ароматических углеводородов. Рекомендуется до приобретения проверять каждую колонку, чтобы удостовериться в том, что она отвечает минимальным требованиям настоящего стандарта в отношении разрешающей способности и селективности.
Новые колонки транспортируют в растворителе, отличном от подвижной фазы, используемой в настоящем стандарте. Поэтому рекомендуется кондиционировать колонку перед ее использованием для текущих анализов путем промывания подвижной фазой (гептаном). Кондиционирование проводят в течение не менее 2 ч при расходе гептана 1 см3/мин, однако иногда необходимо более продолжительное время (до двух дней). В качестве альтернативы можно использовать меньший расход (например, 0,25 см3/мин) в течение не менее 12 ч (например, в течение ночи).
Большая часть колонок, используемых для межлабораторных испытаний, оставалась стабильной длительное время; срок службы колонок может исчисляться двумя годами или более. Однако в работе колонки могут возникнуть небольшие изменения, которые в отсутствие надлежащих мер контроля могут оставаться не замеченными оператором. Лабораториям рекомендуется регулярно регистрировать давление в верхней части колонки и время удерживания эталонов, чтобы иметь возможность следить за работой колонки и системы накопления данных. Настоятельно рекомендуется принимать участие в межлабораторных испытаниях по определению надежности и регулярно использовать эталонные смеси углеводородов в качестве внутреннего стандарта или смеси, утвержденные для оценки методики анализа и колонки.
Колонки, отработавшие свой ресурс и не удовлетворяющие требованиям настоящего стандарта, могут быть регенерированы путем обратного промывания полярным растворителем (например, дихлорметаном при расходе 1 см3/мин в течение 2 ч) с последующим повторным кондиционированием, как в случае новой колонки. Прежде чем удалить отработавшую колонку, рекомендуется тщательно проверить все составные элементы системы с целью выявления утечек, мертвых зон и (или) частичного забивания фильтров, наполнителя колонки, трубок, инжекционных игл, кранов, которые также могут вызвать ухудшение характеристик колонки.
Приложение ДА
(справочное)
Сведения
о соответствии ссылочных европейских региональных стандартов
межгосударственным стандартам
Таблица ДА.1
|
Обозначение европейского регионального |
Степень |
Обозначение и наименование |
|
EN 14214 |
- |
* |
|
EN ISO 1042 (ISO 1042) |
NEQ |
ГОСТ 1770-74 (ИСО 1042-83, ИСО 4788-80) Посуда мерная лабораторная стеклянная. Цилиндры, мензурки, колбы, пробирки. Общие технические условия |
|
EN ISO 3170 (ISO 3170) |
NEQ |
ГОСТ 31873-2012 Нефть и нефтепродукты. Методы ручного отбора проб |
|
EN ISO 3171 (ISO 3171) |
- |
* |
|
* Соответствующий межгосударственный стандарт отсутствует. До его утверждения рекомендуется использовать перевод на русский язык данного европейского стандарта. Примечание - В настоящей таблице использовано следующее условное обозначение степени соответствия стандартов: - NEQ - неэквивалентные стандарты. |
||
Библиография
|
[1] |
EN 590 |
Automotive fuels - Diesel - Requirements and test methods (Топлива для двигателей внутреннего сгорания. Топливо дизельное. Технические требования и методы испытаний) |
|
EN ISO 4259:2006 |
Petroleum products - Determination and application of precision data in relation to methods of test (Нефтепродукты. Определение и применение показателей точности к методам испытаний) |
Ключевые слова: нефтепродукты, ароматические углеводороды, средние дистилляты, метод высокоэффективной жидкостной хроматографии, обнаружение по показателю преломления