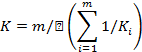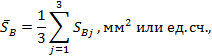ГОСУДАРСТВЕННЫЙ
КОМИТЕТ РОССИЙСКОЙ ФЕДЕРАЦИИ
ПО ОХРАНЕ ОКРУЖАЮЩЕЙ СРЕДЫ
|
|
УТВЕРЖДАЮ
Заместитель
Председателя
Государственного комитета РФ
по охране окружающей среды
_____________
А.А. Соловьянов
24 марта 1999 г.
|
КОЛИЧЕСТВЕННЫЙ
ХИМИЧЕСКИЙ АНАЛИЗ
АТМОСФЕРНОГО ВОЗДУХА И ВЫБРОСОВ В АТМОСФЕРУ
МЕТОДИКА ВЫПОЛНЕНИЯ ИЗМЕРЕНИЙ
МАССОВЫХ КОНЦЕНТРАЦИЙ ПРЕДЕЛЬНЫХ
УГЛЕВОДОРОДОВ С1 - С10 (СУММАРНО, В ПЕРЕСЧЕТЕ
НА УГЛЕРОД), НЕПРЕДЕЛЬНЫХ УГЛЕВОДОРОДОВ
С2 - С5 (СУММАРНО, В ПЕРЕСЧЕТЕ НА УГЛЕРОД) И
АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ
(БЕНЗОЛА, ТОЛУОЛА, ЭТИЛБЕНЗОЛА, КСИЛОЛОВ,
СТИРОЛА) ПРИ ИХ СОВМЕСТНОМ ПРИСУТСТВИИ В
АТМОСФЕРНОМ ВОЗДУХЕ, ВОЗДУХЕ РАБОЧЕЙ ЗОНЫ
И ПРОМЫШЛЕННЫХ ВЫБРОСАХ
МЕТОДОМ ГАЗОВОЙ ХРОМАТОГРАФИИ
ПНД Ф
13.1:2:3.25-99
Методика
допущена для целей государственного экологического
контроля
Москва 1999 г.
(издание 2005 г.)
Методика рассмотрена и одобрена научно-техническим советом
ФГУ «Федеральный центр анализа и оценки техногенного воздействия на окружающую
среду» (ФГУ «ФЦАО»)
Протокол № 2 заседания НТС ФГУ
«ФЦАО» от 4 мая 2005 г.
|
Директор
|
________________
подпись
|
Г.М.
Цветков
|
Разработчики:
Казанское ПНУ «Оргнефтехимзаводы»
ЗАО «ЛЮБЭКОП»
МП «БЕЛИНЭКОМП»
В соответствии с требованиями
ГОСТ
Р ИСО 5725-1-2002 ÷ ГОСТ
Р ИСО 5725-6-2002 и на основании свидетельства о метрологической аттестации
№ 224.02.11.044/2005 в МВИ внесены изменения (Протокол № 2 заседания НТС ФГУ
«ФЦАО» от 04.05.2005).
Настоящая методика предназначена
для измерения массовой концентрации предельных углеводородов С1 - C10
(суммарно, в пересчете на углерод), непредельных углеводородов С2 -
С5 (суммарно, в пересчете на углерод) и ароматических углеводородов
(бензола, толуола, этилбензола, ксилолов, стирола) при их совместном
присутствии в атмосферном воздухе, в воздухе рабочей зоны и в источниках
промышленных выбросов.
|
Наименование
определяемых показателей
|
Диапазон
измерений, мг/м3
|
|
предельные углеводороды С1
- С10 (суммарно, в пересчете на углерод)
|
Oт
0,2 до 1000 вкл.
|
|
непредельные углеводороды С2
- С5 (суммарно, в пересчете на углерод)
|
От
1 до 1000 вкл.
|
|
ароматические углеводороды
(бензол, толуол, этилбензол, ксилол, стирол)
|
От
0,2 до 1000 вкл.
|
Определению не мешают
кислородсодержащие органические соединения.
Настоящая методика обеспечивает получение результатов
измерений с погрешностью, не превышающей значений, приведённых в таблице 1.
Таблица
1 - Значения показателей точности, правильности, повторяемости,
воспроизводимости
|
Показатель
повторяемости (относительное среднеквадратическое отклонение повторяемости),
σr, %
|
Показатель
воспроизводимости (относительное среднеквадратическое отклонение
воспроизводимости), σR, %
|
Показатель
правильности (границы относительной систематической погрешности при
вероятности Р = 0,95), ±δс, %
|
Показатель
точности (границы относительной погрешности при вероятности Р = 0,95),
±δ, %
|
|
4
|
10,5
|
9
|
23
|
Значения показателя точности
методики используют при:
- оформлении результатов анализа, выдаваемых лабораторией;
- оценке деятельности лабораторий на качество проведения
испытаний;
- оценке возможности использования результатов анализа при
реализации методики в конкретной лаборатории.
Газовый хроматограф с пламенно-ионизационным детектором
(предел детектирования по пропану 2,5×10-11 г/с).
Хроматографическая колонка из нержавеющей стали длиной 1
метр и внутренним диаметром 3 мм.
Система обработки данных. (При отсутствии - допускается
применять линейку измерительную, металлическую с ценой деления 1 мм, ГОСТ
427-75; лупу измерительную, ГОСТ
25706-83).
Микрокалькулятор.
Секундомер, кл-3, цена деления 0,2 сек.
Комплект поверочных газовых смесей метан/воздух, ТУ
6-16-2356-92 для градуировки хроматографа (табл. 2):
Таблица 2
|
№ смеси п/п
|
Номер
по реестру ГСО
|
Номинальное
значение и допускаемые отклонения объемной доли масс, концентрации
|
Пределы
допускаемой абсолютной погрешности,
|
|
метана,
млн-1 (%)
|
метана,
|
|
1
|
3896-87
|
7,5±1,0
млн-1
|
|
0,5
|
|
|
5,0
± 0,7
|
0,4
|
|
2
|
3901-87
|
36,0
± 4,0 млн-1
|
|
1,5
|
|
|
24,0
± 3,0
|
1,0
|
|
3
|
3903-87
|
120
± 10 млн-1
|
|
6
|
|
|
80
± 7
|
4
|
|
4
|
4445-88
|
0,08
± 0,01 %
|
|
0,002
|
|
|
530
± 70
|
13
|
|
5
|
4446-88
|
0,20
± 0,02 %
|
|
0,004
|
|
|
1330
± 140
|
30
|
Примечание:
1. Допускается применение
поверочных газовых смесей с другими значениями объемной доли (массовой концентрации)
метана, установленными с относительной погрешностью не более ±8 %.
2.
Значения объемной доли метана, выраженные в млн-1, пересчитываются в
значения массовой концентрации метана мг/м3 (при 20 °С и 101,3 кПа)
путем умножения на 0,667.
ГСО № 5316-90, этилен/азот, объёмная доля этилена 30,0 ± 3,0
млн-1, погрешность ±1,5 млн-1 или ГСО № 5317-90 ,
этилен/азот, объёмная доля этилена 45,0 ± 5,0 млн-1, погрешность
±2,5 млн-1 или эталон сравнения ВНИИМ бензол/азот (воздух) № ЭС 59 с
молярной долей бензола от 30 до 70 млн-1, относительная погрешность
не более ±5 %.
Весы лабораторные типа BЛP-200, ГОСТ
24104-2001.
Аспиратор для отбора проб воздуха, модель 822, ТУ 64-1-862-77.
Пипетки газовые, вместимостью 250 - 500 см3.
Шприцы цельностеклянные, вместимостью 50 - 100 см3,
ТУ 64-1-1279-75.
Термопара хромель-алюмель, с милливольтметром с пределом
измерений до 1000 °С, ГОСТ
9736-81.
Печь муфельная, обеспечивающая нагрев до 1000 °С.
Шкаф сушильный, ТУ 64-1-909-80.
Печь обогрева реактора от газоанализатора ГХЛ-1.
Автотрансформатор лабораторный регулировочного типа ЛАТР-1М,
ТУ 16-671.025-84.
Реактор каталитической очистки газа-носителя из нержавеющей
стали объемом 70 см3 (Приложение Б
, рис. 1).
Фильтрующий патрон для улавливания кислородсодержащих
органических соединений (Приложение В,
рис. 2).
Фильтрующий патрон для улавливания непредельных и
ароматических углеводородов (Приложение Г,
рис. 3).
Набор сит "Физприбор" или сит аналогичного
типа.
Баня водяная, ТУ-64-1-2850-76.
Стекловолокно, ГОСТ 10727-74.
Вата гигроскопическая, ГОСТ 5556-81.
Сетка проволочная.
Эксикатор, ГОСТ
25336-82.
Колба круглодонная типа КГП-3
вместимостью 250 см3, ГОСТ 25336-82.
Посуда лабораторная фарфоровая, ГОСТ 9147-80.
Цветохром ЗК, фр. 0,14 - 0,25 мм и
0,315 - 0,46 мм, ТУ 6-09-26-219-75.
3,3', 3" - Нитрилотрипропионитрил
для хроматографии, ТУ 6-09-06-683-75.
Азотнокислый никель 6-водный, ГОСТ 4055-78.
Магний хлорнокислый 6-водный, ТУ
6-09-2735-73.
Натрий хлористый, ГОСТ 4233-77.
Вода дистиллированная, ГОСТ 6709-72.
Ртуть (I) сернокислая (Получение см.
Приложение А).
Нитрат серебра, ГОСТ 1277-75.
Серная кислота, ГОСТ 4204-77.
Соляная кислота, ГОСТ 3118-77.
Ацетон для хроматографии, ТУ
6-09-1707-77.
Этанол для хроматографии, ТУ
6-09-1710-77.
Гексан для хроматографии, ТУ
6-09-3375-78.
Водород технический, ГОСТ 3022-80, сортность Б.
Воздух, ГОСТ 11882-73.
Шамот, фр. 0,86 - 1,6 мм, ТУ 390-83.
Примечание.
Допускается использование иных средств измерений, вспомогательного оборудования
и реактивов, с метрологическими и техническими характеристиками не хуже, чем у
приведенных выше.
Определение содержания предельных
углеводородов C1 - C10 (суммарно), непредельных
углеводородов С2 - С5 (суммарно) и ароматических
углеводородов (бензола, толуола, этилбензола, ксилолов, стирола) основано на
газохроматографическом разделении компонентов пробы на насадочной колонке,
заполненной 10 % нитрилотрипропионитрила на цветохроме ЗК, с последующей их
регистрацией пламенно-ионизационным детектором. Предельные углеводороды
регистрируются суммарно после улавливания на фильтр непредельных и
ароматических углеводородов. Индивидуальные ароматические углеводороды
регистрируются отдельно. Содержание непредельных углеводородов вычисляется по
разности выходного сигнала, полученного до и после применения фильтрующего
патрона.
4.1 При выполнении анализов необходимо
соблюдать требования техники безопасности при работе с химическими реактивами
по ГОСТ 12.4.007-76.
4.2 Электробезопасность при работе с
электроустановками по ГОСТ 12.1.019-79.
4.3 Организация обучения работников
безопасности труда по ГОСТ 12.0.004-90.
4.4 Помещение лаборатории должно
соответствовать требованиям пожарной безопасности по ГОСТ 12.1.004-91 и иметь средства пожаротушения по ГОСТ 12.4.009-83.
К выполнению измерений и обработке
результатов допускают лиц, имеющих высшее или среднее специальное образование
или опыт работы на газовом хроматографе и в химической лаборатории, прошедших
соответствующий инструктаж, освоивших метод в процессе тренировки и уложившихся
в нормативы оперативного контроля при выполнении процедур контроля погрешности.
При проведении измерений в лаборатории
должны быть соблюдены следующие условия:
Температура воздуха 20 ± 5 °С;
Атмосферное давление 84,0 - 106,7 кПа
(630 - 800 мм рт. ст.);
Влажность воздуха не более 80 % при
температуре 25 °С;
Напряжение в сети 220 ±22 В;
Частота переменного тока 50 ± 1 Гц.
При выполнении измерений должны
соблюдаться следующие условия хроматографического анализа:
|
Колонка заполнена сорбентом на
цветохроме ЗК, фр. 0,14 - 0,25 мм
|
- 10 % нитрилотрипропионитрила
|
|
Наполнитель фильтрующего патрона для
улавливания кислородсодержащих органических соединений
|
Магний хлорнокислый фр. 0,25 - 0,50
мм
|
|
Наполнитель фильтрующего патрона для
улавливания непредельных и ароматических углеводородов
|
Шамот, обработанный раствором
сернокислой закиси ртути; шамот, обработанный раствором AgNО3 в
серной кислоте
|
|
Катализатор для очистки газаносителя
|
12,5 % окиси никеля на цветохроме
ЗК, фр. 0,315 - 0,46 мм
|
|
Температура термостата колонок, °С
|
90
|
|
Температура детектора, °С
|
100
|
|
Температура реактора, °С
|
600
|
|
Газ-носитель
|
каталитически очищенный воздух
|
|
Расход газа-носителя, см3/мин
|
25
|
|
Расход водорода, см3/мин
|
30
|
|
Расход воздуха, см3/мин
|
300
|
|
Объем вводимой пробы, см3
|
1
|
|
Скорость движения ленты
потенциометра, мм/ч
|
600
|
|
Время хроматографического анализа,
мин
|
10
|
|
Отношение выходного сигнала
хроматографа к шуму должно быть не менее
|
10:1 (При отсутствии системы
обработки данных минимальная высота пика - 10 % от шкалы показывающего
прибора) при минимальном рабочем значении масштаба ослабления выходного
сигнала.
|
|
Относительные времена удерживания
определяемых веществ (ориентировочные)
|
Приведены в табл. 3.
|
Предельные углеводороды C1
- C10 и непредельные С2 - С5 элюируются
суммарным пиком до бензола.
Таблица 3 - Ориентировочные относительные времена удерживания
углеводородов
|
Углеводороды
|
Относительные
времена удерживания
|
|
Алифатические
углеводороды С1 - С10
|
0,31
- 0,62 ± 0,05
|
|
Бензол
|
1,00
± 0,15
|
|
Толуол
|
1,45
± 0,2
|
|
Этилбензол
|
1,64
± 0,3
|
|
м-
+ п- Ксилолы
|
2,22
± 0,4
|
|
о-
Ксилол
|
2,92
± 0,5
|
|
Стирол
|
3,98
± 0,7
|
Типовые хроматограммы разделения
углеводородов, полученные при указанных условиях, с использованием фильтрующего
патрона и без него, приведены на рис. 4 и 5 (Приложения Д и Е).
Эффективность разделительной колонки признаётся
удовлетворительной, если степень разрешения (Rs) двух выходящих друг за другом
компонентов (бензола и толуола) не менее 1,5.
Степень
разрешения вычисляют по формуле:
где t1 и t2
- времена удерживания бензола и толуола, с;
μ1, μ2 - ширина пиков
бензола и толуола на половине их высоты, с. При наличии системы обработки
данных μ1 и μ2 можно рассчитать по
формулам:
где h1, h1
и S1, S2 - высоты и площади пиков бензола и
толуола, соответственно.
При нарушении указанного условия необходимо провести
регенерацию хроматографической колонки согласно п. 7.6.
Примечания:
1. Наполнитель
фильтрующего патрона для улавливания кислородсодержащих органических соединений
меняют через каждые 30 анализов.
2.
Наполнитель фильтрующего патрона для улавливания непредельных и ароматических
углеводородов меняют через каждые 10 анализов.
7.1 Приготовление катализатора для очистки газа-носителя
от органических веществ
Взвешивают с точностью до первого десятичного знака 70,0
см3 цветохрома ЗК фракции 0,315 - 0,46 мм, помещают в фарфоровую
чашку и заливают 75,0 - 80,0 см3 водного раствора азотнокислого
никеля из расчета 12,5 % окиси никеля к весу носителя.
Содержимое чашки выпаривают досуха на электрической
плитке при постоянном перемешивании. Высушенный катализатор помещают в
муфельную печь, установленную в вытяжном шкафу. В течение 2-х часов постепенно
поднимают температуру до 600 °С и выдерживают при этой температуре 3 часа до
полного удаления оксидов азота. Подготовленный катализатор засыпают в реактор,
концы которого закрывают тампоном из стекловаты.
Реактор устанавливают в электропечь от прибора ГХЛ-1.
Подачу на печь напряжения, необходимого для поддержания
температуры реактора до 600 °С, осуществляют через автотрансформатор. Контроль
за температурой реактора осуществляют термопарой с милливольтметром.
7.2 Приготовление наполнителя фильтрующего патрона для
улавливания органических примесей, относящихся к классу альдегидов, кетонов,
спиртов, карбоновых кислот п других кислородсодержащих органических соединений
Кристаллогидрат Mg(ClO4)2∙6H2O нагревают в сушильном шкафу в фарфоровой чашке до
температуры 150 - 160 °С. При этой температуре кристаллы плавятся в
кристаллизационной воде. По мере удаления воды жидкость затвердевает в пористую
массу Mg(ClO4)2∙3Н2O. Во время затвердевания препарат
необходимо перемешивать стеклянной палочкой.
Затем температуру повышают до 200 °С (но не выше 230 °С)
и расплавленную соль при этой температуре выдерживают 2 часа. После охлаждения
наполнитель измельчают в фарфоровой ступке, отсеивают фракцию 0,25 - 0,50 мм и
заполняют фильтрующий патрон (Приложение В,
рис. 2).
7.3 Приготовление сорбента для улавливания непредельных
и ароматических углеводородов1
_________________
1 Допускается использовать готовые поглотительные
порошки от газоанализатора УГ-2.
Растворяют 4 г сульфата закиси ртути в 20 см3
х.ч. серной кислоты при нагревании и перемешивании. После полного растворения
навески в фарфоровую чашку вносят 50 г шамота, тщательно перемешивают до
равномерного смачивания зерен и высушивают на плитке при постоянном
перемешивании 15 - 20 мин.
В процессе высушивания обильно выделяются пары серной
кислоты. Высушенный сорбент насыпают в склянку с хорошо пришлифованной пробкой
и оставляют в эксикаторе над серной кислотой. После остывания сорбент
пересыпают в ампулы, которые запаивают.
Затем взвешивают 3 г AgNO3 и растворяют в 54,3 см3 серной кислоты плотностью 1,84 г/см3.
Обрабатывают шамот приготовленным 3 %-ным раствором AgNО3 в серной
кислоте из расчета на 1 г сорбента 0,2 см3 раствора.
Приготовленный таким образом сорбент перемешивают и
помещают в ампулы, которые запаивают на горелке.
Приготовленными сорбентами заполняют стеклянный
фильтрующий патрон (Приложение Г, рис.
3). В узкий конец стеклянной трубки диаметром 5 мм вкладывают небольшой ватный
тампон длиной 5 мм и через широкий конец трубки, соединенной встык с воронкой,
в вертикальном положении при легком и постоянном постукивании штырьком о стенки
трубки насыпают до второй перетяжки слой шамота, обработанного раствором AgNО3
в серной кислоте, третью и четвертую оливы заполняют шамотом, обработанным
раствором сернокислой закиси ртути. Снимают воронку, вкладывают кусочек
гигроскопической ваты слоем 5 мм и немедленно закрывают заглушками оба конца
патрона.
Фильтрующий патрон и вскрытые ампулы с оставшимися
поглотительными порошками должны быстро закрываться заглушками и храниться в
эксикаторе над серной кислотой.
7.4 Приготовление сорбента
Цветохром ЗК фракции 0,14 - 0,25 мм обрабатывают
концентрированной соляной кислотой в течение 3 часов.
После обработки цветохром отмывают дистиллированной водой
до отрицательной реакции на ионы хлора (контролируют по азотнокислому серебру).
Обработанный носитель сушат при 200 °С в течение 3-х
часов, прокаливают при 1000 °С 4 - 5 часов.
7.5 Приготовление насадки
Для приготовления насадки взвешивают с точностью до
первого десятичного знака 30,0 см3 сорбента и рассчитанное
количество (10 % от массы твердого носителя) неподвижной фазы
нитрилотрипропионитрила с точностью до второго десятичного знака. Навеску
неподвижной жидкой фазы растворяют в 35,0 - 40,0 см3 ацетона.
В круглодонную колбу засыпают носитель и туда же вносят растворенную
неподвижную фазу. Объем растворителя должен быть таким, чтобы над поверхностью
твердого носителя образовался слой раствора не более 5 мм. Выдерживают в
течение часа. Растворитель выпаривают в вытяжном шкафу при осторожном
перемешивании содержимого колбы и нагревании на водяной бане до тех пор, пока
насадка не станет сыпучей. Окончательно насадку высушивают на воздухе до
исчезновения запаха растворителя.
7.6 Подготовка хроматографической
колонки
Хроматографическую колонку промывают последовательно
водой, этанолом, гексаном, высушивают в токе воздуха и заполняют сорбентом.
Подготовленную колонку подсоединяют к хроматографу и кондиционируют в токе
газа-носителя при температуре 90 °С в течение 8 часов. После этого колонку
подсоединяют к детектору и кондиционируют ее при температуре 90 °С до
стабилизации нулевой линии при максимальной чувствительности прибора.
7.7 Подготовка хроматографа
Подключение хроматографа к сети, проверка на
герметичность и вывод на режим выполняют согласно инструкции по монтажу и
эксплуатации хроматографа.
Методика предусматривает следующие изменения в газовой
схеме хроматографа (Приложение Ж, рис.
6):
- установку каталитического реактора для очистки
газа-носителя (воздуха) от органических примесей перед блоком подготовки газов;
- исключение фильтра в линии газа-носителя во избежание
накопления органических примесей на нем, дающих фоновое загрязнение;
- исключение вентиля тонкой регулировки на линии
газа-носителя в блоке подготовки газов для устранения изменения давления в
системе при вводе пробы в хроматограф;
- применение крана-дозатора с тефлоновыми уплотнительными
элементами;
- введение фильтрующего патрона для улавливания
органических примесей, относящихся к классу альдегидов, кетонов, спиртов, карбо
новых кислот, простых и сложных эфиров (т.е. кислородсодержащих органических
соединений);
- введение фильтрующего патрона для улавливания
непредельных и ароматических углеводородов.
7.8 Градуировка хроматографа
Градуировку хроматографа для диапазона измеряемых
концентраций от 0,2 до 1000 мг/м3 проводят методом абсолютной
калибровки, используя серию градуировочных смесей с различным содержанием
метана (см. п. 2 табл. 2). Если
измерения предполагается проводить в более узком диапазоне измеряемых
концентраций, то количество градуировочных смесей может быть уменьшено до трёх.
Например: при измерениях в диапазоне от 0,2 до 150 мг/м3 для
градуировки могут быть применены смеси №№ 1 - 3 из табл. 2. Каждую смесь не менее 3 раз подают в
хроматографическую колонку и на полученных хроматограммах определяют значения
площадей пиков в мм2 или ед. сч.
Полученные данные заносят в таблицу, аналогичную
приведенной ниже.
Таблица 4
При анализе каждой градуировочной смеси проверяют
выполнение следующего условия:
где Simax - максимальная площадь
хроматографического пика, мм2 или ед. сч;
Simin - минимальная площадь хроматографического пика,
мм2 или ед. сч;
 i
- среднее арифметическое площадей пиков.
i
- среднее арифметическое площадей пиков.
При невыполнении условия (4)
анализ градуировочной смеси повторяют. При повторном невыполнении условия (4) выясняют и устраняют причины, приводящие к
невыполнению этого условия.
Приведенное значение площади (SПР)
пика метана в i-ой смеси рассчитывают по формуле:
где i - среднее значение
площади пика метана в i-ой смеси, мм2 или в ед. сч.;
М - масштаб ослабления выходного сигнала.
По полученным данным определяют градуировочные
коэффициенты (K1, .... Ki, ..... Km) по формуле:
Градуировочную характеристику признают удовлетворительной
при выполнении следующего условия:
где Kmax - максимальный из m градуировочных коэффициентов;
Kmin - минимальный из m градуировочных
коэффициентов;
m - число
градуировочных коэффициентов;
K -
средневзвешенное значение градуировочных коэффициентов, рассчитанное по
формуле:
При невыполнении условия (7) выясняют и устраняют причины, приводящие к
невыполнению этого условия. После чего повторяют процедуру построения
градуировочной характеристики.
В процессе градуировки измеряют атмосферное давление (Ргр).
Контроль стабильности градуировочной характеристики
проводят не реже одного раза в день, используя одну из газовых смесей,
применяемых при градуировке хроматографа.
Градуировочную характеристику считают
стабильной при выполнении следующего условия:
где (Sпр)к значение приведенной площади пика,
рассчитанное при контроле стабильности градуировочной характеристики, мм3
или ед. сч.;
(Sпр)гр
- значение приведенной площади пика, рассчитанное при построении градуировочной
характеристики, мм2 или ед. сч.;
z - поправочный коэффициент, (см. раздел 10).
При невыполнении условия (9)
выясняют и устраняют причины, приводящие к нестабильности градуировочной
характеристики, и повторяют процедуру контроля стабильности градуировочной
характеристики. При повторном невыполнении условия (9)
строят новую градуировочную характеристику.
Отбор проб следует проводить в соответствии с ГОСТ Р 50820-95 «Оборудование газоочистное и
пылеулавливающее. Методы определения запыленности газопылевых потоков», ПНД Ф 12.1.1-99 «Методические рекомендации по
отбору проб при определении концентрации вредных веществ (газов и паров) в
выбросах промышленных предприятий». Для проб воздуха рабочей зоны - ГОСТ 12.1.005-88 «ССБТ. Общие
санитарно-гигиенические требования к воздуху рабочей зоны» при установившемся
технологическом режиме работы обследуемого источника выделения загрязняющих
веществ в атмосферу.
Анализируемый газ отбирают в
стеклянные газовые пипетки на 250 - 500 см3 с зажимами на концах или
в цельностеклянные шприцы на 50 - 100 см3 с зажимом. Анализируемым
газом промывают пипетку в течение 2 - 3 мин. со скоростью 0,5 - 2 дм3/мин,
перекрывают оба зажима одновременно, выключают аспиратор и отсоединяют пипетку
от системы.
При отборе проб промвыбросов,
находящихся под разрежением, необходимо следить за тем, чтобы в отобранную
пробу не попал воздух. В процессе отбора измеряется температура и давление
(разрежение) газовой пробы у пипетки.
Срок хранения проб не более 5 ч.
Газовые пипетки или цельностеклянные
шприцы с анализируемыми пробами предварительно выдерживают в помещении до
комнатной температуры.
Определение предельных углеводородов в
смеси с непредельными углеводородами проводят путем двойного анализа. Для этого
пробу воздуха вытесняют в дозу крана-дозатора через фильтрующий патрон,
улавливающий непредельные и ароматические углеводороды, и без патрона. Типовые
хроматограммы компонентов воздуха, полученные при указанных условиях, с
использованием фильтрующего патрона и без него, приведены на рис. 4 и 5
(Приложения Д и Е).
Патрон с хлорнокислым магнием для улавливания
кислородсодержащих соединений находится в схеме постоянно.
Ввод пробы в хроматограф осуществляют краном-дозатором не
менее 3 раз. Кран-дозатор переводят в положение «отбор», подсоединяют шприц или
газовую пипетку и вытесняют пробу (в объеме 20 - 30 см3) в дозу.
Объем пробы, вводимой в хроматограф, 1 см3. Вытеснение проб из
пипеток осуществляют насыщенным раствором хлористого натрия. Затем отсоединяют
шприц или газовую пипетку от крана-дозатора для выравнивания в нём давления и
через 1 - 2 сек переводят кран-дозатор в положение «анализ». Подсоединение и
отсоединение шприца (пипетки) необходимо осуществлять таким образом, чтобы в
пробу не попал воздух.
В процессе анализа измеряют атмосферное давление (Ра).
При необходимости в процессе анализа можно менять масштаб
ослабления выходного сигнала.
Для каждой
пробы вычисляют среднее значение площади пика для каждого компонента (В) по
формуле:
где SBj - площади хроматографических пиков,
для которых выполняется условие (4). При
невыполнении условия (4) выясняют и
устраняют причины, приводящие к невыполнению условия (4). После чего процедуру, описанную в разделе 9, повторяют.
Для каждой пробы вычисляют значение приведенной площади
пика SBПР (мм2 или ед. сч.) по формуле
(5) раздела 7.8.
Массовую концентрацию предельных (в
пересчёте на углерод) и ароматических углеводородов вычисляют по формуле:
где: K - градуировочный коэффициент,  или
или 
АВ - коэффициент относительной чувствительности для
вещества "В" (табл. 5);
z - поправочный
коэффициент, учитывающий различия в атмосферном давлении при градуировке и при
анализе. Коэффициент вычисляется по формуле:
где Ргр и Ра
- атмосферное давление при градуировке и при анализе, кПа;
f - коэффициент для приведения значений массовой
концентрации к температуре, соответствующей принятым нормальным условиям.
При анализе воздуха рабочей зоны f = 1,00;
результат измерений приведён к температуре 20 °С (293 К) и давлению 101,3 кПа.
При анализе атмосферного воздуха и выбросов f = 293/273 = 1,07;
результат измерений приведён к температуре 0 °С (273 К) и давлению 101,3 кПа.
Массовую концентрацию непредельных
углеводородов вычисляют по формуле:
где М - масштаб ослабления
выходного сигнала;
AH - коэффициент
относительной чувствительности для непредельных углеводородов (табл. 5);
Sп+н - площадь
пика суммы предельных и непредельных углеводородов на хроматограмме без
патрона, мм2 или в ед. сч;
Sп - площадь пика предельных углеводородов на
хроматограмме с патроном, мм2 или в ед. сч.
Примечание.
Значения коэффициентов относительной чувствительности предельных и непредельных
углеводородов указаны с учетом пересчёта на углерод. Для пересчёта применялась
формула:
где
Су - массовая концентрация углерода, мг/м3;
n -
количество атомов углерода в молекуле;
Мг - относительная
молекулярная масса углеводорода;
С - массовая
концентрация метана, мг/м3.
Таблица 5 - Коэффициенты относительной
чувствительности для пламенно-ионизационного детектора
|
Углеводороды
|
Коэффициенты относительной
чувствительности (А)
|
|
Метан
|
1,00
|
|
Предельные углеводороды
|
0,75
|
|
C1 - C10
|
|
|
(в пересчете на С)
|
0,92
|
|
(в пересчете на пропан)
|
|
|
Этилен
|
0,88
|
|
Пропилен
|
0,88
|
|
Бутилены
|
0,88
|
|
Амилены
|
0,88
|
|
Дивинил
|
0,85
|
|
Изопрен
|
0,85
|
|
Бензол
|
0,81
|
|
Толуол
|
0,83
|
|
Этилбензол
|
0,83
|
|
о-, м-, п- Ксилолы
|
0,83
|
|
Стирол
|
0,81
|
Расхождение между результатами
измерений, полученными в двух лабораториях, не должно превышать предела
воспроизводимости. При выполнении этого условия приемлемы оба результата
измерения, и в качестве окончательного может быть использовано их среднее арифметическое
значение.
Значение предела воспроизводимости (R, %) при
вероятности Р = 0,95 для всех компонентов равно 29,0.
При превышении предела воспроизводимости могут быть использованы
методы проверки приемлемости результатов измерений согласно раздела 5 ГОСТ Р ИСО 5725-6.
Результат измерения в документах,
предусматривающих его использование, может быть представлен в виде: X ±
Δ, Р = 0,95, где Δ - показатель точности методики.
Значение Δ рассчитывают по формуле:
Δ = 0,23Х.
Допустимо результат измерения в
документах, выдаваемых лабораторией, представлять в виде: X ± Δл,
Р = 0,95, при условии Δл < Δ, где
X - результат измерения, полученный в
соответствии с прописью методики;
±Δл - значение
характеристики погрешности результатов измерений, установленное при реализации
методики в лаборатории, и обеспечиваемое контролем стабильности результатов
измерений.
Контроль качества результатов
измерений при реализации методики в лаборатории предусматривает:
- оперативный контроль процедуры
измерений (на основе оценки погрешности при реализации отдельно взятой
контрольной процедуры);
- контроль стабильности результатов
измерений (на основе контроля стабильности среднеквадратического отклонения
повторяемости, среднеквадратического отклонения внутрилабораторной
прецизионности, погрешности).
Алгоритм оперативного контроля
процедуры измерений с применением образцов для контроля
Образцами для контроля являются
эталоны сравнения - газовые смеси этилен/азот и бензол/азот (воздух).
Оперативный контроль процедуры
измерений проводят путем сравнения результата отдельно взятой контрольной
процедуры Kк с нормативом контроля K.
Результат контрольной процедуры Kк рассчитывают по формуле:
где Xк - результат измерений массовой концентрации определяемого
компонента в образце для контроля, мг/м3, рассчитанный по формуле (11) методики;
Фк - объемная или молярная доля этилена (бензола) в
образце для контроля, млн-1;
j - коэффициент пересчета значений объемной (молярной)
доли (млн-1) в значения массовой концентрации (мг/м3) при
20 °С и 101,3 кПа. Для этилена j
= 1,16; для бензола j = 3,24.
Норматив контроля K рассчитывают по формуле
K = Δл,
где ±Δл -
характеристика погрешности результатов измерений, соответствующая
аттестованному значению образца для контроля. Δл =0,01δлjФк, δл - относительное значение
характеристики погрешности результатов измерений.
Примечание
- Допустимо характеристику погрешности результатов измерений при внедрении
методики в лаборатории устанавливать на основе выражения: Δл =
0,84Δ, с последующим уточнением по мере накопления информации в процессе
контроля стабильности результатов измерений.
Процедуру измерений признают
удовлетворительной, при выполнении условия:
При невыполнении условия (16) контрольную процедуру повторяют. При
повторном невыполнении условия (16)
выясняют причины, приводящие к неудовлетворительным результатам, и принимают
меры по их устранению.
Периодичность оперативного контроля процедуры измерений,
а также реализуемые процедуры контроля стабильности результатов измерений
регламентируют в Руководстве по качеству лаборатории.
Приготовление ртути (I) сернокислой
(Ю.В.
Карякин, И.И. Ангелов. Чистые химические вещества. Москва. 1974 г. стр. 312)
1. Препарат
реактивной чистоты можно получить растворением ртути в концентрированной серной
кислоте:
2Hg + 2H2SО4
= Hg2SО4↓ + SО2↑ + 2H2О
Нагревают (под тягой) 1 вес.ч.
ртути с 0,5 - 1 вес ч. H2SО4 (пл. 1,84 г/см3),
пока приблизительно половина ртути не перейдет в твердую соль. Неиспользованную
ртуть сливают. Соль промывают небольшим количеством холодной воды и сушат.
2.
Сернокислую ртуть можно получить, осаждая ее серной кислотой из раствора соли
Hg22+.
Hg2(NО3)2 + H2SO4 = Hg2SO4↓ + 2HNО3
В раствор Hg2(NO3)2
прибавляют разбавленную серную кислоту (1:2) до полного осаждения Hg2SО4.
Осадок промывают небольшим количеством воды и сушат.
РЕАКТОР КАТАЛИТИЧЕСКОЙ ОЧИСТКИ
ГАЗА-НОСИТЕЛЯ
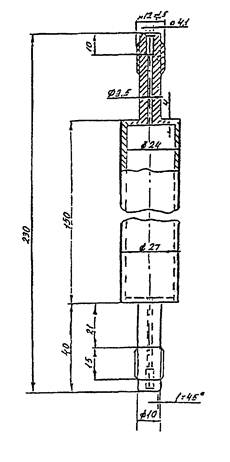
Рис. 1
УСТАНОВКА ФИЛЬТРУЮЩЕГО ПАТРОНА ДЛЯ
УЛАВЛИВАНИЯ КИСЛОРОДСОДЕРЖАЩИХ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ

1 - стенка термостата; 2 - переходник;
3 - стеклянный патрон; 4 - гайка;
5 - прокладка; 6 - гайка; 7 - прокладка; 8 - соединительная трубка; 9 - гайка.
Рис. 2
ФИЛЬТРУЮЩИЙ ПАТРОН ДЛЯ УЛАВЛИВАНИЯ КИСЛОРОДСОДЕРЖАЩИХ ОРГАНИЧЕСКИХ
СОЕДИНЕНИЙ
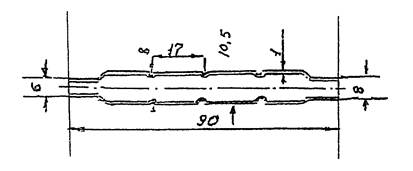
Рис. 3
ТИПОВАЯ ХРОМАТОГРАММА РАЗДЕЛЕНИЯ
ИСКУССТВЕННОЙ СМЕСИ УГЛЕВОДОРОДОВ И КИСЛОРОДСОДЕРЖАЩИХ СОЕДИНЕНИЙ НА КОЛОНКЕ С
НИТРИЛОТРИПИОНИТРИЛОМ
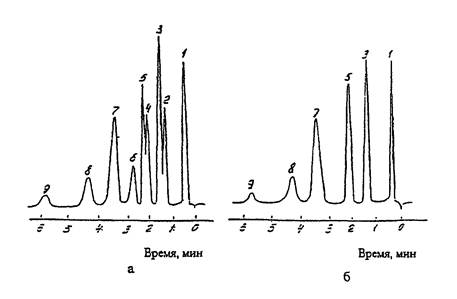
а - анализ без фильтрующего патрона
б - анализ с фильтрующим патроном
1 - гексан; 2
- этилацетат; 3 - бензол + ацетон; 4 - метилэтилкетон;
5 - толуол; 6 - бутилацетат; 7 - m, n - ксилолы; 8 - о-ксилол; 9 - стирол.
Рис. 4
ТИПОВАЯ ХРОМАТОГРАММА РАЗДЕЛЕНИЯ
ИСКУССТВЕННОЙ СМЕСИ ПРЕДЕЛЬНЫХ, НЕПРЕДЕЛЬНЫХ И АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ НА
КОЛОНКЕ С НИТРИЛОТРИПИОНИТРИЛОМ

а - анализ без фильтрующего патрона: 1
- гексан + непредельные углеводороды;
2 - бензол; 3 - толуол; 4 - m, n - ксилолы; 5 - о-ксилол; 6 - стирол.
б - анализ с фильтрующим патроном: 1 - гексан.
Рис. 5
ПРИНЦИПИАЛЬНАЯ СХЕМА ГАЗОВОЙ ОБВЯЗКИ ХРОМАТОГРАФА

1, 2 - баллоны со сжатым воздухом и
водородом соответственно; 3, 4 - редукторы;
5 - печь прибора ГХЛ-1; 6 - реактор; 7 - блок подготовки газов; 8 - регулятор
давления;
9 -дроссель; 10 - фильтр; 11 - блок анализатора; 12 - фильтрующий патрон для
улавливания непредельных и ароматических углеводородов; 13 - кран-дозатор;
14 - фильтрующий патрон для улавливания кислородсодержащих соединений;
15 - хроматографическая колонка; 16 - детектор; 17 - усилитель; 18 -
регистратор.
Рис. 6
СОДЕРЖАНИЕ