ФЕДЕРАЛЬНОЕ АГЕНТСТВО
ПО ТЕХНИЧЕСКОМУ РЕГУЛИРОВАНИЮ И МЕТРОЛОГИИ
|
|
НАЦИОНАЛЬНЫЙ |
ГОСТ
Р |

НЕФТЬ СЫРАЯ
Газохроматографический
метод определения
распределения компонентов по диапазону
температур кипения
|
|
Москва Стандартинформ 2011 |
Предисловие
Цели и принципы стандартизации в Российской Федерации установлены Федеральным законом от 27 декабря 2002 г. № 184-ФЗ «О техническом регулировании», а правила применения национальных стандартов Российской Федерации - ГОСТ Р 1.0-2004 «Стандартизация в Российской Федерации. Основные положения»
Сведения о стандарте
1 ПОДГОТОВЛЕН Открытым акционерным обществом «Всероссийский научно-исследовательский институт по переработке нефти» (ОАО «ВНИИ НП») на основе собственного аутентичного перевода на русский язык стандарта, указанного в пункте 4
2 ВНЕСЕН Техническим комитетом по стандартизации ТК 31 «Нефтяные топлива и смазочные материалы»
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 27 декабря 2010 г. № 1132-ст
4 Настоящий стандарт идентичен стандарту АСТМ Д 5307-97(2007) «Определение пределов выкипания сырой нефти методом газовой хроматографии» (ASTM D 5307-97(2007) «Standard test method for determination of boiling range distribution of crude petroleum by gas chromatography»).
Наименование настоящего стандарта изменено относительно наименования указанного стандарта для приведения в соответствие с ГОСТ Р 1.5-2004 (подраздел 3.5).
При применении настоящего стандарта рекомендуется использовать вместо ссылочных стандартов АСТМ соответствующие им национальные стандарты Российской Федерации и межгосударственные стандарты, сведения о которых приведены в дополнительном приложении ДА
5 ВВЕДЕН ВПЕРВЫЕ
Информация об изменениях к настоящему стандарту публикуется в ежегодно издаваемом информационном указателе «Национальные стандарты», а текст изменений и поправок - в ежемесячно издаваемых информационных указателях «Национальные стандарты». В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ежемесячно издаваемом информационном указателе «Национальные стандарты». Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет
СОДЕРЖАНИЕ
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
НЕФТЬ СЫРАЯ
Газохроматографический
метод определения распределения компонентов
по диапазону температур кипения
Crude petroleum. Gas chromatography method for determination of components boiling temperature range distribution
Дата введения - 2012-07-01
1 Область применения
1.1 Настоящий стандарт распространяется на свободную от воды сырую нефть и устанавливает метод определения распределения компонентов нефти по температурам кипения до 538 °С (1000 Т) включительно. Материал, кипящий при температуре выше 538 °С, регистрируют как остаток. Настоящий метод испытания применим к представительным образцам сырой нефти, пробы которой можно растворить в растворителе и отобрать микрошприцем.
1.2 Значения, установленные в единицах СИ, следует считать стандартными. Единицы (дюйм, фунт), приведенные в скобках, даны только для сведения.
1.3 Применение настоящего стандарта может быть связано с использованием опасных веществ, операций и оборудования. В настоящем стандарте не предусмотрено рассмотрение всех вопросов обеспечения безопасности, связанных с его применением. Пользователь настоящего стандарта несет ответственность за разработку необходимых мер техники безопасности и охраны здоровья персонала, а также определяет целесообразность применения законодательных ограничений перед его использованием.
Особые меры безопасности указаны в 7.2, 7.5, 7.6 и 7.9.
2 Нормативные ссылки
В настоящем стандарте использованы нормативные ссылки на следующие стандарты:
2.1 Стандарты АСТМ1):
_____________
1) По ссылкам на стандарты АСТМ следует обращаться на сайт www.astm.org или в службу АСТМ по электронной почте service@astm.org. За информацией, содержащейся в сборниках стандартов АСТМ, необходимо обращаться на сводную страницу документации стандартов, находящуюся на сайте АСТМ.
АСТМ Д 2892 Метод определения фракционного состава сырой нефти (на колонке с 15 теоретическими тарелками) [ASTM D 2892, Test method for distillation of crude petroleum (15-theoretical plate column)]
АСТМ Д 4057 Руководство по ручному отбору проб нефти и нефтепродуктов (ASTM D 4057, Practice for manual sampling of petroleum and petroleum products)
3 Термины и определения
3.1 В настоящем стандарте применены следующие термины с соответствующими определениями:
3.1.1 площадь узкого участка пиков (area slice): Площадь, получаемая в результате интегрирования сигналов хроматографического детектора в пределах определенного интервала значений времени удерживания.
При определении площади узких участков пиков (6.2.2) параметры детектирования пика выделяют об водкой и записывают интегрированный сигнал детектора как площадь узких участков пиков для последовательных фиксированных по длительности интервалов времени.
3.1.2 скорректированная площадь участков пиков (corrected area slice): Площадь узких участков пиков, скорректированная на нулевую линию, полученная вычитанием соответствующей площади участка пиков в опыте с образцом и в холостом (без образца) опыте. Может потребоваться корректировка отклонения сигнала.
3.1.3 совокупная скорректированная площадь (cumulative corrected area): Общая сумма скорректированных площадей узких участков пиков от начала анализа до определенного времени удерживания без учета площади пиков, не принадлежащих анализируемому образцу (например, растворителю).
3.1.4 температура начала кипения; ТНК (initial boiling point, IBP): Температура (соответствующая времени удерживания), при которой совокупная скорректированная площадь равна 0,5 % теоретической общей площади.
3.1.5 остаток (residue, RES): Количество образца, кипящее при температуре выше 538 °С (1000 °F).
3.1.6 теоретическая общая площадь T (theoretical total area, T): Площадь, которая была бы получена, если бы из колонки был элюирован весь образец.
Расчет величин, определенных выше, приведен в 12.3.
3.2 Обозначения
3.2.1 Углеводородные соединения обозначают числом атомов углерода в соединении. Префикс используют для указания формы углеводородной цепи, а подстрочное число обозначает количество атомов углерода (например, нормальный декан обозначают n-С10; изотетрадекан - i-С14).
4 Сущность метода
4.1 Образец сырой нефти разбавляют дисульфидом углерода и полученный раствор вводят в газохроматографическую колонку, разделяющую углеводороды в соответствии со значениями температуры кипения. Температуру колонки повышают с воспроизводимой линейной скоростью и в течение всего анализа регистрируют площадь, занятую хроматограммой.
Температуру кипения определяют по оси времени путем сопоставления с калибровочной кривой, полученной в таких же хроматографических условиях при анализе смеси н-парафинов с известной температурой кипения до температуры 538 °С (1000 °F) включительно. Остаток, кипящий при температуре выше 538 °С, определяют повторным анализом сырой нефти, к которой добавлен внутренний стандарт. Исходя из этих данных, рассчитывают распределение компонентов по фракционному составу образца, свободного от воды.
5 Назначение и применение
5.1 Определение распределения компонентов по диапазонам выкипания является важным требованием в анализе сырой нефти. Данную информацию можно использовать для приблизительного подсчета производительности нефтеперерабатывающего завода и, наряду с другой информацией, для оценки экономики использования одной конкретной нефти взамен другой.
5.2 Результаты, полученные настоящим методом испытания, эквивалентны результатам, полученным по методу испытания АСТМ Д 2892 (приложение XI).
5.3 Настоящий метод испытания быстрее метода испытания по АСТМ Д 2892, и его можно использовать, когда в наличии только малые объемы образцов. Также настоящий метод дает результаты вплоть до 538 °С, в то время как метод по АСТМ Д 2892 ограничен температурой 400 °С.
6 Аппаратура
6.1 Газовый хроматограф
Можно использовать любой газовый хроматограф с нижеуказанными характеристиками, отвечающий техническим требованиям по эксплуатации, приведенным в разделе 10.
6.1.1 Детектор
В настоящем методе испытания используют только пламенно-ионизационный детектор (ПИД).
Детектор должен обеспечивать непрерывную работу при максимальной или даже более высокой температуре для используемой колонки. Детектор должен быть подсоединен к колонке таким образом, чтобы избежать возможной конденсации.
6.1.2 Программатор температуры колонки
Хроматограф должен работать при воспроизводимом линейном программировании температуры в диапазоне, достаточном для установления времени удерживания не менее 1 мин при начальной температуре кипения и элюирования соединений вплоть до температуры 538 °С (1000 °F), прежде чем будет достигнут конец температурной программы.
6.1.3 Криогенный термостат колонки
Если начальная температура кипения сырой нефти ниже 90 °С (194 °F), то начальная температура колонки должна быть ниже температуры окружающей среды. Это требует применения на газовом хроматографе криогенного охлаждения.
В таблице 1 приведены типичные начальные температуры колонки.
Таблица 1 - Типичные рабочие условия
|
Колонка |
|||
|
1 |
2 |
3 |
|
|
Длина колонки, мм (дюйм) |
457 (18) |
610 (24) |
457 (18) |
|
Диаметр колонки, мм (дюйм) |
3,17 (1/8) |
3,17 (1/8) |
3,17 (1/8) |
|
Жидкая фаза |
10 % UCW-982 |
3 % OV-1 |
10 % SE-30 |
|
Материал носителя |
Хромосорб PA)-AW |
Хромосорб WA)-HP |
Хромосорб PA)-AW |
|
Значение начальной температуры колонки, °С |
-30 |
-30 |
-40 |
|
Значение конечной температуры кипения, °С |
380 |
350 |
360 |
|
программируемая скорость изменения температуры, °С/мин |
10 |
10 |
10 |
|
Тип газа-носителя |
Азот |
Гелий |
Азот |
|
Скорость потока газа-носителя, мл/мин |
25 |
20 |
28 |
|
Температура детектора, °С |
400 |
380 |
400 |
|
Температура отверстия для ввода пробы |
380 |
375 |
400 |
|
А) Использовался в межлабораторном совместном исследовании 8 лабораториями для 5 образцов. |
|||
6.1.4 Система ввода образца
Можно использовать одну из следующих систем ввода образца.
6.1.4.1 Мгновенное испарение
Испарительная система ввода образца должна обеспечивать непрерывную работу при температуре, близкой или равной максимальной используемой температуре колонки.
Система ввода образца также должна быть подсоединена к хроматографической колонке таким образом, чтобы избежать конденсации образца.
6.1.4.2 Во время эксплуатации жидкий образец вводят непосредственно в головную часть колонки. Должны быть предусмотрены устройства для программирования температуры всей колонки, включая температуру ввода образца, вплоть до максимальной используемой колонкой температуры.
6.1.5 Регулятор расхода
Хроматограф должен быть снабжен регулятором расхода, поддерживающим поток газа-носителя постоянным по всему рабочему температурному диапазону колонки ± 1 %. Входное давление газа-носителя, подаваемое к хроматографу, должно быть достаточно высоким, чтобы компенсировать увеличение противодавления в колонке, по мере программирования повышения температуры. Обнаружено, что давление на входе, равное 550 кПа (80 psig), является достаточным для колонок по таблице 1.
6.2 Система получения данных
6.2.1 Самописец
Для графического представления сигнала ПИД используют самопишущий потенциометр диапазоном измерения от 0 до 1 мВ с временем отклика по полной шкале 2 с или менее.
Обычный интегратор или компьютерная система обработки данных хроматографии должна использоваться для интегрирования и накопления сигнала детектора. Система интегратор - компьютер должна иметь обычное хроматографическое программное обеспечение для измерения времени удерживания и площадей пиков элюирования (режим обнаружения пика). Дополнительно система должна обеспечивать преобразование непрерывно интегрируемого сигнала детектора в площади узких участков, представляющих смежные временные интервалы фиксированной продолжительности (режим определения площади узкого участка). Рекомендуемый временной интервал составляет 1 с, но не должен превышать 12 с.
Система должна обеспечивать вычитание площади узкого участка из площади соответствующего пика анализируемого образца. Альтернативно хроматограмму нулевой линии можно вычесть из хроматограммы образца, и чистую результирующую хроматограмму обработать в режиме разбивки на узкие участки.
Также используют компьютерную программу, которая выполняет расчет узких участков после проведения анализа.
6.3 Колонка
Можно использовать любую газохроматографическую колонку, обеспечивающую разделение в зависимости от температур кипения и отвечающую техническим характеристикам, указанным в разделе 10. В таблице 1 приведены успешно применяемые колонки и условия проведения испытания.
6.4 Микрошприц
Для введения образца используют микрошприц вместительностью 5 или 10 мкл. Для отбора пробы жидкости должны применяться автоматические устройства.
7 Реактивы и материалы
7.1 Чистота реактивов
Химические вещества квалификации х.ч. следует применять при всех испытаниях. Если нет других указаний, все реактивы должны соответствовать спецификациям Американского химического общества, где они имеются в наличии. Можно использовать реактивы другой квалификации при условии, что чистота реактива установлена и достаточно высокая, что обеспечивает точность определения при его использовании.
7.2 Воздух должен быть нулевого класса (т.е. свободный от углеводородов) для применения в ПИД.
Предупреждение - Сжатый воздух - это газ под высоким давлением, поддерживающий горение.
7.3 Хлорид кальция безводный (CaCl2).
7.4 Калибровочная смесь - смесь н-парафинов, растворенных в дисульфиде углерода (см. 7.5, предупреждение), с пределом кипения образца до 538 °С (1000 °F) включительно.
Не менее чем одно соединение смеси должно иметь температуру кипения, равную начальной температуре кипения образца или ниже. При необходимости к калибровочной смеси можно добавить метан, этан, пропан или бутан, вводя газовым шприцем 1 см3 чистого газообразного соединения в запечатанную колпачком с перегородкой виалу, содержащую остальную калибровочную смесь. Если пики н-парафинов можно четко (недвусмысленно) идентифицировать на хроматограмме образца, то их значения времени удерживания можно использовать для калибровки.
7.5 Дисульфид углерода (CS2) с массовой долей основного вещества не менее 99 %. Используется в качестве растворителя, уменьшающего вязкость, так как он смешивается с сырой нефтью и плохо определяется пламенно-ионизационным детектором.
Предупреждение - Дисульфид углерода - чрезвычайно летучее, воспламеняемое и токсичное вещество.
Предупреждение - Гелий и азот являются сжатыми газами под высоким давлением.
Предупреждение - н-Октан воспламеняем и вреден при вдыхании.
7.8 Смесь для испытания отклика детектора. Точно взвешенная смесь приблизительно равных масс не менее 6 н-парафинов, охватывающая число атомов углерода в диапазоне от 10 до 44. Растворяют одну часть этой смеси приблизительно в пяти частях CS2 (или в достаточном количестве CS2, чтобы обеспечить стабильный раствор при комнатной температуре).
Предупреждение - Водород под давлением - чрезвычайно воспламеняющийся газ.
7.10 Внутренний стандарт. Смесь приблизительно равных количеств четырех н-парафинов с н-С14 по н-С17 включительно. Концентрации отдельных компонентов могут быть точно неизвестны, но они должны быть в пределах линейного диапазона используемой электронной системы/детектора.
7.11 Жидкая фаза. Инертная неполярная жидкость или смола низкой летучести.
Обычно используют силиконовую смолистую резину. Наиболее удовлетворительна загрузка жидкой фазы - от 3 % до 10 %.
7.12 Твердый носитель. Диатомит (инфузорная земля) или равноценный инертный материал в виде частиц. Обычные размеры частиц находятся в диапазонах 60/80 меш или 80/100 меш.
8 Отбор проб
8.1 Пробы для анализа по настоящему методу необходимо отбирать в соответствии с АСТМ Д 4057 или аналогичными национальными стандартами.
8.2 Пробы должны храниться в герметичных емкостях.
9 Подготовка аппаратуры
9.1 Подготовка колонки
Может быть использован любой подходящий метод, который соответствует требованиям к колонке, указанным в разделе 10.
9.2 Кондиционирование колонки
Колонку необходимо кондиционировать при максимальной температуре, чтобы уменьшить дрейф нулевой линии, вызываемый утечкой наполнителя колонки. Колонку можно кондиционировать очень быстро и эффективно при использовании следующей процедуры.
9.2.1 Подсоединяют колонку к системе ввода, оставив свободным конец для детектора.
9.2.2 Продувают колонку газом-носителем при температуре окружающей среды.
9.2.3 Закрывают кран газа-носителя и дают колонке полностью сбросить давление.
9.2.4 Герметично закрывают открытый конец для детектора подходящим способом (колпачком).
9.2.5 Повышают температуру колонки до максимальной рабочей температуры и поддерживают колонку при этой температуре в течение 4 - 6 ч, не пропуская поток через колонку.
9.2.6 Охлаждают колонку до температуры окружающей среды.
9.2.7 Удаляют колпачок с колонки и подсоединяют колонку к детектору. Вновь устанавливают поток носителя.
9.2.8 Программируют несколько раз температуру колонки до максимальной с обычной скоростью потока газа-носителя.
9.3 Альтернативным методом кондиционирования колонок является метод продувки колонки (отсоединенной от детектора) газом-носителем в течение 12 - 16 ч при максимальной рабочей температуре колонки. Этот метод является эффективным для колонок с первоначальной загрузкой 5 % жидкой фазы.
9.4 Хроматограф
Работу на приборе проводят в соответствии с инструкцией изготовителя. Типичные рабочие условия приведены в таблице 1.
9.4.1 Для нормальной работы колонки как газожидкостной хроматографической и чтобы исключить возможность застывания жидкой фазы, следует избегать очень низкой начальной температуры колонки. За дополнительной инструкцией следует обращаться к производителю жидкой фазы. Начальная температура колонки должна быть низкой настолько, чтобы получить калибровочную кривую, отвечающую требованиям, указанным в 6.1.3.
9.4.2 Двуокись кремния от сжигания материала колонки откладывается на горелке ПИД. Это отложение необходимо регулярно удалять, очищая щеткой, во избежание изменения характеристик чувствительности детектора.
9.4.3 Отложения двуокиси кремния могут привести к тому, что пламя погаснет. Эту проблему можно решить, используя горелку внешним диаметром не менее 0,76 мм (0,030 дюйма).
10 Характеристики системы
10.1 Разрешающая способность
Анализируют аликвоту смеси для испытания разрешающей способности колонки (7.7), используя те же условия, что и при анализе образцов. Разрешающая способность колонки для пиков парафинов н-С16 и н-C18 должна находиться между тремя и десятью при расчете по следующей формуле (см. рисунок 1)
![]() (1)
(1)
где R - разрешающая способность;
t2 - время выхода максимума пика н-С18, с;
t1 - время выхода максимума пика н-С16, с;
Y2 - ширина на половине высоты пика н-С18, с;
Y1 - ширина на половине высоты пика н-С16, с.
10.2 Повторяемость времени удерживания
Система должна удовлетворять повторяемости времени удерживания при испытании калибровочной смеси. Время удерживания пиков для калибровочной смеси не должно превышать 6 с.
10.3 Проверка работы системы
Анализируют чувствительность детектора (7.8), используя калибровочную смесь в условиях идентичных анализу образцов. Рассчитывают коэффициенты чувствительности относительно н-декана по следующей формуле
Fn = (Cn/An)/(C10/A10), (2)
где Fn - коэффициент чувствительности относительно н-декана;
Сn - концентрация н-парафина в смеси;
Аn - площадь пика н-парафина;
С10 - концентрация н-декана в смеси;
А10 - площадь пика н-декана.
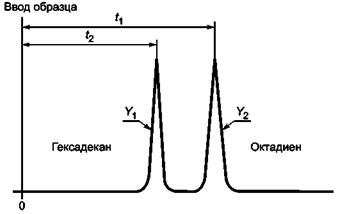
Рисунок 1 - Параметры разрешающей способности колонки
10.3.1 Коэффициент чувствительности Fn каждого н-парафина не должен отклоняться от единицы более чем на 10 %.
10.3.2 Было отмечено, что для некоторых хроматографов коэффициенты чувствительности для высококипящих н-парафинов (от н-С20 до н-С44) изменялись после того, как были проанализированы несколько образцов сырой нефти. Для проверки стабильности системы проверочное испытание повторяют после анализа десяти образцов. Если система все еще отвечает установленному режиму работы (10.3.1), то нет необходимости повторять эту проверку после последующих анализов. Однако полезно повторять проверочные испытания при замене компонентов детектора.
11 Проведение испытания
11.1 Анализ компенсации нулевой линии
Чтобы компенсировать дрейф нулевой линии и сдвиг (отклонение) сигнала, вычитают профиль площади узких участков холостого опыта из анализа образца, чтобы получить узкие участки скорректированной площади. Этот профиль получают следующим образом.
11.1.1 После установки условий, соответствующих техническим требованиям, температуру термостата колонки программируют на достижение максимальной температуры, которая будет использоваться, и выдерживают не менее 10 мин.
11.1.2 Следуя строго установленной процедуре, охлаждают колонку до выбранной начальной температуры и дают в течение не менее 3 мин установиться равновесию при этой температуре.
В точно установленное процедурой время запускают температурную программу колонки, не вводя образец (холостой опыт).
11.1.3 Получают данные по режиму узкого участка площади (6.2.2), регистрируя узкие участки площади для каждого интервала времени от начала анализа до конца. Важно, чтобы время проведения всех измерений было одинаковым как для холостого опыта, так и для испытания образца.
11.1.4 В день проведения анализов обязательно выполняют холостой опыт.
Примечание 1 - Трудно получить полностью удовлетворительную нулевую линию, когда компенсация испарения жидкой фазы проводится равноценными сдвоенными колонками и детекторами. На практике наилучшую компенсацию можно получить, непосредственно вычитая профиль площади холостого опыта из профиля анализа образца с использованием одной колонки.
Примечание 2 - Некоторые заводские газовые хроматографы имеют возможность проводить корректировки нулевой линии (из сохраненного холостого опыта) непосредственно на сигнал детектора. При применении таких систем дополнительная корректировка площади узких участков возможно не потребуется. Однако если к сигналу после компенсации нулевой линии добавляется электронное отклонение, то может потребоваться дополнительная корректировка, то есть вычитание отклонения. Чтобы установить, есть ли эта дополнительная корректировка сигнала, следует обратиться к инструкциям конкретного оборудования.
11.2 Калибровка времени удерживания в зависимости от температуры кипения
11.2.1 В тех же самых условиях, что и для холостого опыта, и следуя тому же строго установленному стандартному режиму (11.1), вводят в хроматограф соответствующую аликвоту калибровочной смеси (7.4)..
Данные регистрируют таким образом, чтобы получить значения времени удерживания и площади пиков для каждого компонента (режим детектирования пика).
11.2.1.1 Объем вводимой калибровочной смеси должен быть таким, чтобы избежать искажения форм пика любого компонента в результате перегрузки колонки. Искаженные пики приведут в результате к смещению верхушек пиков (т.е. к ошибочным значениям времени удерживания) и следовательно к ошибкам в определении температур кипения. Количество жидкой фазы в колонке имеет прямое отношение к величине образца.
11.2.2 Строят график, нанося время удерживания для пика каждого компонента напротив соответствующей ему температуры (точки) кипения, как представлено на рисунке 2. Температуры кипения н-парафинов приведены в таблице 2.
Составляют таблицу по этим данным и сохраняют их для последующих расчетов.
11.2.3 Калибровочная кривая должна быть фактически линейным графиком зависимости температуры кипения от времени удерживания. Поскольку нереально управлять колонкой так, чтобы полностью ликвидировать изгиб на нижнем конце кривой, калибровочная смесь должна содержать не менее одного н-парафина с температурой кипения, равной ТНК образца или ниже.
Более точной является экстраполяция кривой на верхнем конце (538 °С) при условии, что экстраполяция не проводится вне программируемого участка температуры. Однако для большей точности следует принимать во внимание крайние точки на калибровочной кривой как в верхней, так и в нижней части. Если нормальные парафины можно однозначно идентифицировать в образце, то эти значения времени удерживания можно использовать для калибровки.
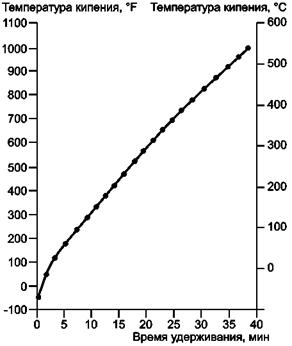
Рисунок 2 - Типичная калибровочная кривая
Таблица 2 - Температура кипения нормальных парафинов1)
_____________
1) Green, L.E., «Chromatograph Gives Boiling Point», Hydrocarbon Processing, May 1976, p. 205.
|
Температура кипения |
Число атомов углерода |
Температура кипения |
|||
|
°С |
°F |
°С |
°F |
||
|
1 |
-162 |
-259 |
23 |
380 |
716 |
|
2 |
-89 |
-127 |
24 |
391 |
736 |
|
3 |
-42 |
-44 |
25 |
402 |
756 |
|
4 |
0 |
32 |
26 |
412 |
774 |
|
5 |
36 |
97 |
27 |
422 |
792 |
|
6 |
69 |
156 |
28 |
431 |
808 |
|
7 |
98 |
209 |
29 |
440 |
825 |
|
8 |
126 |
258 |
30 |
449 |
840 |
|
9 |
151 |
303 |
31 |
458 |
856 |
|
10 |
174 |
345 |
32 |
466 |
871 |
|
11 |
196 |
385 |
33 |
474 |
885 |
|
12 |
216 |
421 |
34 |
481 |
898 |
|
13 |
235 |
455 |
35 |
489 |
912 |
|
14 |
254 |
489 |
36 |
496 |
925 |
|
15 |
271 |
520 |
37 |
503 |
937 |
|
16 |
287 |
549 |
38 |
509 |
948 |
|
17 |
302 |
576 |
39 |
516 |
961 |
|
18 |
316 |
601 |
40 |
522 |
972 |
|
19 |
330 |
626 |
41 |
528 |
982 |
|
20 |
344 |
651 |
42 |
534 |
993 |
|
21 |
356 |
674 |
43 |
540 |
1004 |
|
22 |
369 |
685 |
44 |
545 |
1013 |
11.2.4 Калибровку «время удерживания - температура кипения» обязательно проводят в день выполнения анализов.
11.3 Подготовка образца
11.3.1 Образцы, содержащие очень легкие компоненты, хранят при температуре от 0 °С до минус 5 °С. Перед открытием закрытый образец оставляют при этой температуре не менее 4 ч (предпочтительно на ночь).
11.3.2 Для обеспечения гомогенности образец встряхивают или перемешивают и наливают небольшую порцию (около 100 см3) образца для последующего взвешивания и анализа.
11.3.3 Тяжелую вязкую сырую нефть следует нагреть и перемешать, чтобы обеспечить гомогенность.
11.3.4 Поскольку воду не измеряют с помощью ПИД, то перед взвешиванием образца его необходимо высушить (обезводить).
В виалу вместимостью 50 см3 добавляют 2 - 3 г осушающего средства, например безводного хлорида кальция, и наполовину заполняют ее образцом.
Виалу плотно закрывают крышкой и энергично встряхивают. Дают смеси выстояться несколько минут для оседания агента.
Пипеткой удаляют осушенный слой сырой нефти для взвешивания и анализа образца.
11.3.5 Взвешивают не менее 10 г осушенного образца с точностью 0,1 мг в виалу вместимостью 25 см3.
11.3.6 В ту же виалу добавляют примерно 1 г смеси внутреннего стандарта. Определяют массу с точностью 0,1 мг.
11.3.7 Смесь разбавляют примерно равным объемом дисульфида углерода.
11.3.8 Виалу плотно закрывают крышкой, и смесь энергично встряхивают в течение 3 мин или до тех пор, пока компоненты смеси полностью не растворятся друг в друге (солюбилизируются).
Этот раствор используют для анализа «сырая нефть плюс внутренний стандарт» (11.4.1).
11.3.9 Во второй виале такое же количество обезвоженного образца, как и в 11.3.5, растворяют в равном объеме дисульфида углерода. Этот раствор используют для анализа «сырая нефть без внутреннего стандарта» (11.4.4).
11.4 Анализ образца
11.4.1 В условиях точно таких же, как и для холостого опыта и анализа калибровки (11.1 и 11.2), следуя строго определенным условиям анализа (11.1), в хроматограф вводят 1 мкл разбавленной смеси «сырая нефть плюс внутренний стандарт». Регистрируют профиль площади каждого интервала времени до конца анализа (включительно).
11.4.2 Продолжают анализ до достижения времени удерживания, эквивалентного температуре кипения 538 °С (1000 °F).
При этой температуре прекращают регистрацию площадей узких участков на хроматограмме.
11.4.3 Для максимального удаления тяжелых компонентов, остающихся в колонке, продолжают нагревание колонки до тех пор, пока сигнал ПИД не вернется к нулевой линии. Чтобы ускорить этот процесс можно повысить температуру колонки.
11.4.4 Колонку охлаждают до исходной температуры. Используя условия, идентичные 11.4.1, вводят 1 мкл образца «сырая нефть без внутреннего стандарта» (11.3.9). Регистрируют профиль площади каждого интервала времени до конца анализа (включительно).
11.4.5 Испытание образца как с внутренним стандартом (11.4.1), так и без него (11.4.4) можно выполнять в любом порядке.
12 Обработка результатов
12.1 Корректировка площади
12.1.1 Скорректированные площади узких участков пиков для каждого испытания (11.4.1 и 11.4.4) рассчитывают, вычитая соответствующие площади узких участков профиля холостого опыта (11.1) из каждого результата обоих испытаний, указанных выше (примечание 2).
12.1.2 Суммируют скорректированные площади узких участков для каждого испытания, чтобы получить в течение испытания общую скорректированную площадь в конце каждого временного интервала.
12.2 Теоретическая общая площадь (рисунок 3)
12.2.1 На основании времени удерживания из калибровочной хроматограммы (11.2.1) выбирают значения времени удерживания, которые на 5 % меньше времени удерживания н-С14, и одно, которое на 5 % больше времени удерживания н-С7.
Эти значения времени определяют сегмент хроматограммы, включающий пики внутреннего стандарта.
Регистрируют общую площадь в рамках этого сегмента из хроматограммы образца «сырая нефть плюс внутренний стандарт» (11.4.1) (площадь AIS, рисунок 3а). Также регистрируют общую площадь того же самого сегмента из хроматограммы образца «сырая нефть» (11.4.4) (площадь BIS, рисунок 3b).
12.2.2 Регистрируют общую площадь обеих хроматограмм до времени удерживания, эквивалентного температуре кипения 538 °С (1000 °F).
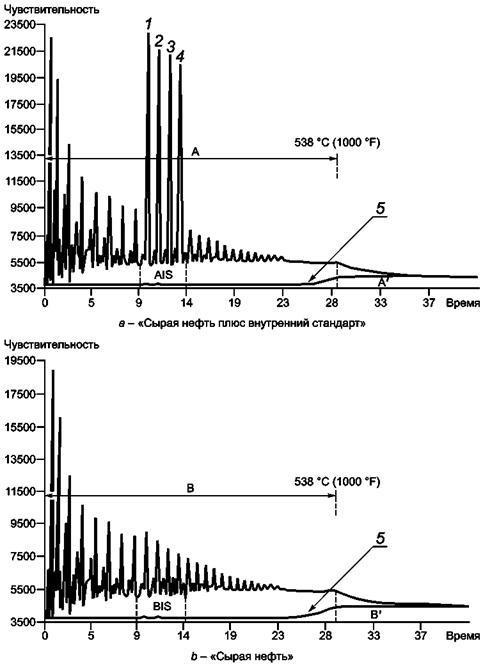
1 - углеводород С14; 2 - углеводород С15;
3 - углеводород С16; 4 - углеводород С17; 5
- нулевая линия;
А и В - общая площадь элюирования вплоть до 538 °С (1000 °F); А и В - площади,
соответствующие
неэлюированному образцу; AIS - площадь «сырая нефть плюс внутренний стандарт»;
BIS - площадь сегмента, где внутренний стандарт элюирует на рисунке 3а
Рисунок 3 - Типичные хроматограммы
12.2.3 Рассчитывают массовую долю W внутреннего стандарта в образце «сырая нефть плюс внутренний стандарт» по формуле
W = I/(S + I), (3)
где I - масса внутреннего стандарта, г;
S - масса образца, г.
12.2.4 Рассчитывают соотношение площадей r вне сегмента внутреннего стандарта до времени удерживания, эквивалентного температуре кипения 538 °С (1000 °F) включительно, хроматограммы образца «сырая нефть» и хроматограммы образца «сырая нефть плюс внутренний стандарт» по формуле
r = (B - BIS)/(A - AIS), (4)
где В - общая площадь до 538 °С (1000 °F) включительно хроматограммы образца «сырая нефть»;
BIS - общая площадь сегмента внутреннего стандарта хроматограммы образца «сырая нефть»;
А - общая площадь до 538 °С (1000 °F) включительно хроматограммы «сырая нефть плюс внутренний стандарт»;
AIS - общая площадь сегмента внутреннего стандарта хроматограммы образца «сырая нефть плюс внутренний стандарт».
12.2.5 Рассчитывают теоретическую общую площадь Т для хроматограммы образца «сырая нефть» (площадь В + В¢, рисунок 3b) по формуле
Т = [(AIS × r) - BIS] [(I - W)/W], (5)
где AIS - общая площадь сегмента внутреннего стандарта хроматограммы образца «сырая нефть плюс внутренний стандарт»;
r - соотношение площадей вне сегмента внутреннего стандарта до 538 °С (1000 °F) (включительно) для обеих хроматограмм [формула (4)];
BIS - общая площадь сегмента внутреннего стандарта хроматограммы образца «сырая нефть»;
W - массовая доля внутреннего стандарта в образце «сырая нефть плюс внутренний стандарт» [формула (3)].
12.2.6 Рассчитывают остаток RES свыше 538 °C (1000 °F), % масс, по формуле
RES = 100 - (В/Т × 100), (6)
где В - общая площадь хроматограммы образца «сырая нефть» до температуры 538 °С (1000 °F);
Т - теоретическая общая площадь хроматограммы образца «сырая нефть» [формула (5)].
12.3 Расчет распределения температур кипения
12.3.1 Регистрируют время, при котором общая площадь в начале хроматограммы образца «сырая нефть» равна 0,5 % теоретической общей площади T [формула (5)]. Температура, эквивалентная этому времени, - начальная температура кипения образца ТНК.
12.3.2 Скорректированную общую площадь в конце каждого интервала времени умножают на 100 и делят на теоретическую общую площадь Т [формула (5)]. В результате получают выход отгона (% масс.) для каждого узкого участка в каждый временной интервал.
12.3.3 Записывают в виде таблицы парами общий выход отгона (% масс.) и время удерживания в каждом временном интервале.
12.3.4 Используя линейную интерполяцию, при необходимости определяют время, соответствующее каждому проценту в интервале, с 1 % до процента, соответствующего времени элюирования при температуре 538 °С (1000 °F).
12.3.5 Для каждого 1 % и соответствующего ему времени удерживания определяют соответствующую температуру по таблице калибровочных данных «время удерживания - температура кипения» (11.2.2).
13 Оформление результатов
13.1 Записывают следующую информацию:
13.1.1 Температуру с точностью до 0,5 °С (1 °F) в точке начала кипения и температуру через каждый 1 %.
13.1.2 Общий остаток при температуре выше 538 °С с точностью до 0,1 %.
14 Прецизионность и отклонение
14.1 Прецизионность
Прецизионность настоящего метода испытания определена межлабораторным статическим исследованием с участием 8 лабораторий на 5 образцах.
14.1.1 Повторяемость
Расхождение между двумя последовательными результатами испытаний, полученными одним и тем же оператором на одном и том же приборе при постоянных условиях на идентичных веществах в течение длительного времени при нормальном и правильном выполнении испытания данным методом, считается правильным, если полученные значения превышают указанные в таблице 3 только в одном случае из двадцати.
14.1.2 Воспроизводимость
Расхождение между двумя единичными независимыми результатами испытаний, полученными разными операторами, работающими в разных лабораториях, на идентичных испытуемых образцах в течение длительного времени при нормальном и правильном выполнении испытания данным методом, считается правильным, если полученные значения превышают указанные в таблице 3 только в одном случае из двадцати.
Таблица 3 - Повторяемость и воспроизводимость
|
Повторяемость, °С |
Воспроизводимость, °С |
|
|
ТНК |
3,7 |
10,6 |
|
5 |
4,7 |
14,8 |
|
10 |
6,9 |
11,3 |
|
20 |
6,8 |
15,4 |
|
30 |
7,6 |
20,4 |
|
40 |
9,3 |
24,6 |
|
50 |
10,6 |
30,3 |
|
60 |
11,8 |
25,9 |
|
70 |
17,6 |
39,2 |
|
80 |
24,8 |
38,8 |
|
85 |
18,8 |
38,8 |
|
90 |
20,7 |
44,9 |
|
Остаток |
2,6 % масс. |
8,1 % масс. |
Примечание 3 - Образцы, включенные в исследование, имели остатки в диапазоне от 3 % до 30 %. Образцы, имеющие остатки вне этого диапазона, могут иметь другую прецизионность.
14.2 Отклонение
Настоящий метод испытания для определения распределения пределов кипения сырой нефти газовой хроматографией не имеет отклонений, так как распределение пределов кипения можно определить только конкретным методом испытания.
14.2.1 Невозможно строго теоретически определить распределение пределов кипения нефтяных фракций из-за сложности смеси, а также неучтенных взаимодействий между компонентами (например, поведение азеотропных смесей). Некоторые другие методы, используемые для определения распределения пределов выкипания, предусматривают использование какого-нибудь физического процесса, например обычной перегонки или газохроматографической идентификации. Это приводит к невозможности установления истинного значения, по отношению к которому можно рассчитать отклонение, так как все определения возможны только для конкретного метода.
Приложение
Х1
(справочное)
Сравнение с традиционной перегонкой
Х1.1 Метод испытания АСТМ Д 2892 является стандартным для традиционной перегонки сырой нефти.
Х1.2 Было проведено сравнение результатов настоящего метода испытания с результатами метода испытания АСТМ Д 2892.
По методу испытания АСТМ Д 2892 трудно установить ТНК и легкую часть сырой нефти, и для предотвращения крекинга образца перегонку необходимо ограничить температурой 400 °С.
Х1.3 Сноска1) особенно важна, поскольку показывает непосредственное сравнение результатов, полученных в круговых испытаниях настоящего метода и метода испытания АСТМ Д 2892. Включены данные пяти лабораторий.
_____________
1) Ceballo, CD., et al Rev. Tee. INTEVEP, 7 (1), pp. 81 - 83.
Приложение ДА
(справочное)
Таблица ДА.1
|
Обозначение |
Степень |
Обозначение и
наименование соответствующего |
|
АСТМ Д 2892 |
- |
* |
|
АСТМ Д 4057 |
MOD |
ГОСТ 2517-85 «Нефть и нефтепродукты. Методы отбора проб» ГОСТ Р 52659-2006 «Нефть и нефтепродукты. Методы ручного отбора проб» |
|
* Соответствующий национальный стандарт отсутствует. До его утверждения рекомендуется использовать перевод на русский язык данного стандарта. Перевод данного стандарта находится в Федеральном информационном фонде технических регламентов и стандартов. Примечание - В настоящей таблице использовано следующее условное обозначение степени соответствия стандартов: MOD - модифицированные стандарты. |
||
Ключевые слова: сырая нефть, газохроматографический метод, нефть, распределение компонентов