|
УТВЕРЖДАЮ Директор ООО «Производственно-экологическое
________ Ю.Г. Василенко 20.05.2004 |
КОЛИЧЕСТВЕННЫЙ ХИМИЧЕСКИЙ АНАЛИЗ ВОД
МЕТОДИКА
ВЫПОЛНЕНИЯ ИЗМЕРЕНИЙ
МАССОВЫХ КОНЦЕНТРАЦИЙ НЕФТЕПРОДУКТОВ И ЖИРОВ
(ПРИ ИХ СОВМЕСТНОМ ПРИСУТСТВИИ)
В ПРОБАХ ПИТЬЕВЫХ, ПРИРОДНЫХ
И ОЧИЩЕННЫХ СТОЧНЫХ ВОД
МЕТОДОМ ИК-СПЕКТРОФОТОМЕТРИИ
ФР.1.31.2006.02410
НОВОСИБИРСК
2004
МВИ разработана
Испытательным аналитическим центром
Новосибирского института органической химии
имени Н.Н. Ворожцова СО РАН
и
Производственно-экологическим предприятием
«СИБЭКОПРИБОР» (г. Новосибирск),
аттестована в соответствии с ГОСТ Р 8.563-96.
Аттестация осуществлена по результатам
метрологической экспертизы материалов по разработке методики
Уральским научно-исследовательским институтом метрологии.
Свидетельство об аттестации методики № 224.01.05.235/2004.
Право тиражирования и реализации МВИ принадлежит
ООО «Производственно-экологическое предприятие «СИБЭКОПРИБОР».
Адрес: 630058, г. Новосибирск, ул. Русская, д. 41
Телефон: (383) 333-74-58, 332-91-36, 333-82-66
Факс: (383) 333-74-58, 332-91-36
1. НАЗНАЧЕНИЕ И ОБЛАСТЬ ПРИМЕНЕНИЯ
Настоящий документ устанавливает ИК - спектрофотометрическую методику количественного химического анализа (КХА) проб питьевых, природных и очищенных сточных вод для определения в них массовой концентрации нефтепродуктов (НП) и жиров (Ж) при их совместном присутствии в диапазоне концентраций: НП - от 0,04 до 5,00 мг/дм3, Ж - от 0,10 до 10,00 мг/дм3.
Неионогенные ПАВ в концентрации менее 0,1 мг/дм3 не мешают определению жиров.
2. МЕТОД ИЗМЕРЕНИЯ
Метод основан на извлечении НП и Ж четыреххлористым углеродом из анализируемой пробы воды (при pH £ 2) посредством двукратной экстракции. Экстракт делят на две приблизительно равные части. В первой части экстракта определяют суммарную концентрацию НП и Ж. Вторую часть экстракта подвергают хроматографическому разделению в колонке, заполненной оксидом алюминия, и в элюате определяют массовую концентрацию НП. По разности результатов этих определений находят массовую концентрацию Ж в анализируемой пробе воды.
Количественное определение веществ проводят по интенсивности поглощения С-Н связей в инфракрасной области спектра (2930 ± 70) см-1.
3. ПРИПИСАННЫЕ ХАРАКТЕРИСТИКИ ПОГРЕШНОСТИ ИЗМЕРЕНИЙ И ЕЕ СОСТАВЛЯЮЩИХ
3.1. Методика выполнения измерений обеспечивает получение результатов измерений с погрешностью, не превышающей значений, приведенных в таблице 1.
Таблица 1 - Диапазон измерений, значения показателей точности, повторяемости, воспроизводимости, правильности методики.
|
Показатель повторяемости (относительное среднеквадратическое отклонение повторяемости), sr, % |
Показатель воспроизводимости (относительное среднеквадратическое отклонение воспроизводимости), sR, % |
Показатель правильности (границы относительной систематической погрешности при вероятности Р = 0,95), ± δc, % |
Показатель точности (границы относительной погрешности при вероятности Р = 0,95), ± δ, % |
|
|
нефтепродукты |
||||
|
От 0,04 до 0,25 вкл. |
10 |
18 |
9 |
37 |
|
Свыше 0,25 до 0,5 вкл. |
9 |
14 |
7 |
28 |
|
Свыше 0,5 до 5,0 вкл. |
8 |
12 |
7 |
25 |
|
жиры |
||||
|
От 0,1 до 0,5 вкл. |
8 |
13 |
18 |
31 |
|
Свыше 0,5 до 10,0 вкл. |
5 |
12 |
10 |
26 |
3.2. Значения показателя точности методики используют при:
- оформлении результатов измерений, выдаваемых лабораторией;
- оценке деятельности лабораторий на качество проведения испытаний;
- оценке возможности использования результатов измерений при реализации методики выполнения измерений в лаборатории.
4. СРЕДСТВА ИЗМЕРЕНИЙ, ВСПОМОГАТЕЛЬНЫЕ УСТРОЙСТВА, РЕАКТИВЫ И МАТЕРИАЛЫ
При выполнении измерений массовой концентрации НП и Ж используют следующие средства измерений, вспомогательные устройства, реактивы и материалы.
4.1. Средства измерений
|
- Концентратомер КН-2, |
ИШВЖ. 004 ТУ |
|
КН-2м |
ИШВЖ. 010 ТУ |
|
- Государственный стандартный образец состава раствора НП в четыреххлористом углероде |
ГСО 7822-2000 |
|
- Государственный стандартный образец состава тристеарина |
ГСО 8126-2002 |
|
- Весы лабораторные ВЛР-200 |
по ГОСТ 24104 |
|
- Меры массы |
по ГОСТ 7328 |
|
- Пипетки вместимостью 5, 10 см3 |
по ГОСТ 29227 |
|
- Цилиндры мерные вместимостью 10, 25, 1000 см3 |
по ГОСТ 1770 |
|
- Колбы мерные 2-50-2 |
по ГОСТ 1770 |
4.2. Вспомогательные устройства
|
- Шкаф сушильный общелабораторный, обеспечивающий поддержание температуры от 105 до 110 °C |
|
|
- Печь муфельная ПМ-8 |
по ТУ 79-337 |
|
- Установка из стекла для перегонки растворителей: |
|
|
- перегонная колба вместимостью 1 дм3 |
|
|
- дефлегматор елочный (длиной не менее 25 см) |
по ГОСТ 25336 |
|
- холодильник ХПТ (длиной не менее 30 см) |
по ГОСТ 25336 |
|
- плитка электрическая с закрытой спиралью |
по ГОСТ 14919 |
|
- термометр лабораторный (от 0 до 100 °C, с ценой деления 0,1 °C) |
по ГОСТ 28498 |
|
- Стаканы химические вместимостью 50 см3 |
по ГОСТ 25336 |
|
- Стаканчик для взвешивания - (бюкс) высокий |
по ГОСТ 25336 |
|
- Воронка лабораторная |
по ГОСТ 25336 |
|
- Экстрактор ЭЛ-1 |
|
|
- Воронка делительная 1,0 дм3 |
по ГОСТ 25336 |
|
- Колонка хроматографическая - стеклянная трубка диаметром 7 мм длиной 200 мм, нижний конец оттянут до диаметра 1 - 2 мм |
|
|
- Штатив для хроматографических колонок |
|
|
- Эксикатор с силикагелем или обезвоженным хлористым кальцием |
по ГОСТ 25336 |
|
- Чашка фарфоровая |
по ГОСТ 9147 |
|
- Стеклянные палочки длиной 12 - 15 см |
|
|
- Шпатель |
|
|
- Стеклянные бутыли вместимостью 1 дм3 с притертой пробкой для отбора и хранения проб. |
|
- Четыреххлористый углерод, х.ч. |
по ГОСТ 20288 |
|
|
или о.с.ч. |
или ТУ 6-09-3219 |
|
|
- Оксид алюминия, для хроматографии |
по ТУ 6-09-3916 |
|
|
или ч.д.а. с размером зерен 0,10 - 0,25 мм (40 - 100 меш.) |
или ГОСТ 8136 |
|
|
- Натрий сернокислый, безводный, ч. |
по ГОСТ 4166 |
|
|
- Кислота серная |
по ГОСТ 4204 |
|
|
- Кислота азотная |
по ГОСТ 4461 |
|
|
- Вода дистиллированная |
по ГОСТ 6709 |
|
|
- Стекловолокно |
по ГОСТ 10727 |
|
|
или стекловата |
||
|
- Бумага универсальная индикаторная для измерения pH |
||
Допускается использование реактивов, изготовленных по другой нормативно-технической документации, в том числе импортных, с квалификацией не ниже указанной в 4.3.
5. ТРЕБОВАНИЯ БЕЗОПАСНОСТИ И ОХРАНЫ ОКРУЖАЮЩЕЙ СРЕДЫ
5.1. При выполнении анализов необходимо соблюдать требования техники безопасности при работе с химическими реактивами по ГОСТ 12.1.007.
5.2. Электробезопасность при работе с электроустановками соблюдается по ГОСТ 12.1.019.
5.3. Помещение лаборатории должно соответствовать требованиям пожарной безопасности по ГОСТ 12.1.004 и иметь средства пожаротушения по ГОСТ 12.4.009. Содержание вредных веществ не должно превышать допустимых концентраций по ГОСТ 12.1.005.
5.4. Организация обучения работающих безопасности труда проводится по ГОСТ 12.0.004.
6. ТРЕБОВАНИЯ К КВАЛИФИКАЦИИ ОПЕРАТОРОВ
К выполнению измерений и обработке их результатов допускают специалиста, имеющего высшее или среднее специальное химическое образование или опыт работы в химической лаборатории, прошедшего соответствующий инструктаж, освоившего метод в процессе тренировки и уложившегося в нормативы оперативного контроля процедуры анализа.
7. УСЛОВИЯ ИЗМЕРЕНИЙ
Условия окружающей среды, при которых обеспечивается требуемая точность измерений, следующие:
|
- температура окружающего воздуха, °C |
20 ± 5; |
|
- атмосферное давление, кПа (мм рт. ст.) |
97,3 - 104,6 (730 - 780); |
|
- относительная влажность воздуха |
не более 80 % (при t° = 25 °C); |
|
- частота переменного тока, Гц |
50 ± 1; |
|
- напряжение питания электросети, В |
220 ± 22. |
8. ОТБОР ПРОБ
Общие требования к отбору проб - по ГОСТ Р 51592 и ГОСТ Р 51593. При отборе должен быть исключен захват пленки с поверхности воды.
Пробы воды отбирают в отожженные стеклянные бутыли, предварительно ополоснутые отбираемой водой. Отобранные пробы не фильтруют и не делят.
Рекомендуемый отбираемый объем пробы в зависимости от предполагаемого содержания НП и Ж в воде должен соответствовать значениям, указанным в таблице 2.
Таблица 2 - Объем проб воды
|
Объем пробы, дм3 |
|
|
нефтепродукты |
|
|
От 0,04 до 1,0 вкл. |
1,00 ± 0,10 |
|
Свыше 1,0 до 5,0 вкл. |
0,50 ± 0,05 |
|
жиры |
|
|
От 0,1 до 1,0 вкл. |
1,00 ± 0,10 |
|
Свыше 1,0 до 10,0 вкл. |
0,50 ± 0,05 |
Экстракцию проводят не позднее 3 часов после отбора пробы. Сорбированные на стенках бутыли НП и Ж должны быть смыты растворителем, который затем используют для экстракции. При невозможности проведения экстракции в течение этого срока пробу консервируют добавлением смеси серной кислоты и четыреххлористого углерода из расчета 1 см3 концентрированной серной кислоты и 2,0 - 3,0 см3 четыреххлористого углерода на 1 дм3 пробы. При экстракции эти объемы следует учитывать. Допускается добавление консервантов в пустую емкость до отбора пробы. Законсервированные пробы можно хранить при температуре 3 - 5 °C не более 72 часов.
При отборе проб составляют сопроводительный документ, в котором указывают:
- цель анализа, предполагаемые загрязнители,
- место, время отбора,
- номер пробы,
- должность, фамилия отбирающего пробу, дата.
9. ПОДГОТОВКА К ВЫПОЛНЕНИЮ ИЗМЕРЕНИЙ
9.1. Подготовка посуды
При выполнении измерений массовой концентрации НП и Ж необходимо тщательно соблюдать чистоту химической посуды.
Используемую посуду следует тщательно вымыть и ополоснуть не менее двух раз дистиллированной водой и высушить. Для мытья химической посуды разрешается использовать концентрированные серную и азотную кислоты. Запрещается использовать для мытья все виды синтетических моющих средств!
Рекомендуется иметь отдельный набор посуды, который используется только для определения НП и Ж. Категорически запрещается смазывать шлифы и краны делительных воронок всеми видами смазок!
9.2. Подготовка реактивов и материалов
9.2.1. Подготовка четыреххлористого углерода
Проверяют чистоту каждой партии четыреххлористого углерода в соответствии с руководством по эксплуатации концентратомера КН-2, КН-2м. В кювету заливают чистый четыреххлористый углерод и помещают в прибор. На табло появится цифровое показание, характеризующее чистоту четыреххлористого углерода. Если это показание лежит в пределах от - 10,0 до 20,0 мг/дм3, то четыреххлористый углерод пригоден для работы. В противном случае выполняют очистку растворителя следующим образом.
В экстрактор или делительную воронку вместимостью 1 дм3 помещают около 0,4 дм3 четыреххлористого углерода, добавляют 0,5 дм3 дистиллированной воды и перемешивают в течение 1 минуты. Слой четыреххлористого углерода сливают в колбу. Процедуру повторяют с новой порцией дистиллированной воды.
К промытому четыреххлористому углероду добавляют около 10 г безводного сульфата натрия и, периодически перемешивая, выдерживают 10 - 15 минут. Обезвоженный четыреххлористый углерод декантируют в перегонную колбу и перегоняют при температурном интервале от 76 до 78 °C, собирая отдельно первые 50 - 60 см3 (затем отбрасывают), основную фракцию (собственно очищенный четыреххлористый углерод) и оставляя в перегонной колбе около 50 см3 четыреххлористого углерода.
Для приготовления растворов по 9.3, проведения калибровки прибора по 9.5, а также разбавления анализируемого раствора при измерении концентраций по 10.3 следует использовать четыреххлористый углерод, дополнительно пропущенный через хроматографическую колонку с оксидом алюминия, подготовленным по 9.2.2.
9.2.2. Подготовка оксида алюминия 2-ой степени активности
Сорбент просеивают через сито и используют мелкую фракцию. Оксид алюминия прокаливают в фарфоровой или кварцевой чашке в муфельной печи при температуре 600 °C в течение 4 часов, остужают до 150 °C, после чего помещают в эксикатор и охлаждают до комнатной температуры.
Если при прокаливании оксид алюминия приобретает желтый цвет, то он непригоден для использования. Срок хранения прокаленного оксида алюминия в плотно закрытой таре составляет один месяц.
Перед использованием необходимое количество прокаленного оксида алюминия взвешивают, добавляют 3 % (по массе) дистиллированной воды, плотно закрывают, встряхивают несколько минут и выдерживают в течение суток при комнатной температуре.
9.2.3. Подготовка безводного сульфата натрия
Перед употреблением безводный сульфат натрия высушивают при температуре 105 - 110 °C в течение 8 часов в сушильном шкафу, охлаждают и хранят в эксикаторе. Срок хранения составляет 1 месяц.
9.2.4. Подготовка стекловолокна или стекловаты
Стекловолокно или стекловату выдерживают в разбавленной (1:1) серной или азотной кислоте в течение 12 часов, промывают водопроводной, затем дистиллированной водой и сушат в сушильном шкафу.
Примечание - Допускается использование ваты медицинской по ГОСТ 5556 (хлопковой, не синтетической!). Перед использованием вату тщательно промывают четыреххлористым углеродом и высушивают при комнатной температуре.
9.2.5. Подготовка хроматографических колонок
В нижнюю часть вымытой и высушенной колонки помещают комочек стекловолокна или стекловаты. Затем в колонку засыпают 3 г оксида алюминия (подготовленного по 9.2.2) и вновь помещают слой стекловолокна или стекловаты (0,5 см). Оксид алюминия используют в колонке однократно.
9.2.6. Регенерация четыреххлористого углерода
Сливы четыреххлористого углерода, образующиеся в процессе подготовки прибора к работе, ополаскивания посуды при подготовке и в ходе определения, а также после анализа проб, собирают в склянку для слива. При накоплении достаточного количества сливов выполняют очистку растворителя одним из следующих методов:
- в соответствии с «Руководством по проведению адсорбционной очистки отходов четыреххлористого углерода»;
- после осушки сульфатом натрия перегоняют, собирая среднюю фракцию.*
___________
* Сливы четыреххлористого углерода, содержащие ГСО НП (углеводородов), перегонке не подлежат!
Проверяют чистоту получаемого четыреххлористого углерода аналогично 9.2.1 и, в случае необходимости, повторяют очистку.
9.2.7. Подготовка животного жира
Животный жир используется в случае отсутствия ГСО 8126-2002 состава тристеарина.
Навеску 5 г животного жира (лучше говяжьего) растворяют в 50 см3 четыреххлористого углерода. Полученный раствор фильтруют через слой стекловолокна или стекловаты (подготовленных по 9.2.4), а затем упаривают. Полученный очищенный жир используют для приготовления растворов по 9.3 аналогично ГСО.
Основной раствор НП в четыреххлористом углероде готовят из ГСО 7822-2000 состава раствора НП (углеводородов) в четыреххлористом углероде**. Для этого ГСО состава раствора НП из ампулы количественно переносят в мерную колбу вместимостью 50 см3, затем ампулу тщательно промывают 5 раз четыреххлористым углеродом порциями по 3 см3, сливая в мерную колбу, и затем доводят объем раствора в колбе до метки четыреххлористым углеродом. Раствор перемешивают и хранят в холодильнике при температуре 0 - 5 °C не более 6 месяцев. Перед использованием раствор выдерживают при комнатной температуре не менее 30 минут.
___________
** Допускается использование ГСО состава раствора НП (углеводородов) в четыреххлористом углероде другого типа с аналогичными метрологическими характеристиками. В этом случае основной раствор готовят в соответствии с инструкцией по применению используемого ГСО.
Массовая концентрация НП в полученном растворе составляет 1000 мг/дм3.
Относительная погрешность приготовления - 0,6 %.
Основной раствор тристеарина в четыреххлористом углероде: 50 мг ГСО 8126-2000 состава тристеарина помещают в мерную колбу вместимостью 50 см3 растворяют в четыреххлористом углероде, затем доводят объем раствора в колбе до метки четыреххлористым углеродом, перемешивают.
Основной раствор можно хранить при температуре 0 - 5 °C в течение 6 месяцев. Перед использованием раствор выдерживают при комнатной температуре не менее 30 минут.
Массовая концентрация тристерина в полученном растворе составляет 1000 мг/дм3. Относительная погрешность приготовления - 2,5 %.
Основные растворы НП и тристеарина используют в качестве добавки при контроле качества результатов измерений по 15.
Рабочий раствор НП в четыреххлористом углероде готовят разбавлением основного раствора НП. Для этого в мерную колбу вместимостью 50 см3 вносят пипеткой 5,0 см3 основного раствора НП и доводят объем раствора в колбе до метки четыреххлористым углеродом. Раствор перемешивают и хранят в холодильнике при температуре 0 - 5 °C не более 2 месяцев. Перед использованием раствор выдерживают при комнатной температуре не менее 30 минут.
Массовая концентрация НП в полученном растворе составляет 100 мг/дм3. Относительная погрешность приготовления - 0,7 %.
Градуировочные растворы НП в четыреххлористом углероде готовят непосредственно перед использованием путем разбавления рабочего раствора НП. Для этого в мерные колбы вместимостью 50 см3 вносят пипеткой последовательно 1,0; 2,5; 5,0; 10,0; 25,0 см3 рабочего раствора НП и доводят объемы растворов в колбах до метки четыреххлористым углеродом. Растворы тщательно перемешивают.
Массовая концентрация НП в полученных растворов составляет 2, 5, 10, 20, 50 мг/дм3 соответственно. Относительная погрешность приготовления не превышает 2,5 %. Перед использованием раствор выдерживают при комнатной температуре не менее 30 минут.
Градуировочные растворы тристеарина в четыреххлористом углероде готовят непосредственно перед использованием путем разбавления основного раствора тристеарина. Для этого в мерные колбы вместимостью 50 см3 последовательно помещают 0,5; 1,5; 2,5; 4; 5 см3 основного раствора тристеарина и доводят до метки четыреххлористым углеродом.
Массовая концентрация тристеарина в полученных растворах составляет 10, 30, 50, 80, 100 мг/дм3 соответственно.
Относительная погрешность δ (Р = 0,95) градуировочных растворов тристеарина, обусловленная процедурой приготовления, не превышает 3,5 %. Растворы можно хранить при температуре 0 - 5 °C в течение 3 месяцев. Перед использованием раствор выдерживают при комнатной температуре не менее 30 минут.
Градуировочные растворы используют для контроля работоспособности концентратомера в области измеряемых значений массовых концентраций НП и Ж.
9.4. Подготовка и использование концентратомера
Подготовку концентратомера к работе и его использование осуществляют в соответствии с руководством по эксплуатации.
10. ВЫПОЛНЕНИЕ ИЗМЕРЕНИЙ
10.1. Экстракция
Пробу анализируемой воды полностью переносят в делительную воронку соответствующей вместимости, приливают разбавленную (1:9) серную кислоту до pH £ 2 (контролируют по индикаторной бумаге). Если проба воды была предварительно законсервирована в соответствии с 8, то серную кислоту не добавляют. Сосуд, в котором находилась проба, тщательно ополаскивают 5 см3 четыреххлористого углерода и выливают растворитель в делительную воронку. Добавляют туда еще 5 см3 четыреххлористого углерода (с учетом консервации по 8 общий объем четыреххлористого углерода должен быть 10 см3). Выполняют экстракцию, встряхивая делительную воронку не менее 10 минут, затем отстаивают в течение 10 минут. После расслоения фаз нижний слой (экстракт) сливают в колбу. Повторяют экстракцию с новой порцией четыреххлористого углерода объемом 10 см3. Затем экстракты объединяют и подвергают обработке по 10.2 или оставляют на хранение. Экстракт допускается хранить в течение 1 недели при температуре 3 - 4 °C. Объем анализируемой пробы воды измеряют мерным цилиндром.
Допускается выполнять экстракцию в экстракторах, входящих в комплект прибора. В этом случае руководствуются соответствующими инструкциями по эксплуатации. При проведении экстракции в экстракторе необходимо следить, чтобы экстрагент равномерно распределился по всей пробе воды, скорость вращения мешалки должна равняться либо превышать 2500 об./мин., время экстракции - 5 мин.
Экстракт сушат безводным сульфатом натрия (не менее 4 г), подготовленным по 9.2.3, в течение 10 минут, добавляя его в стаканчик небольшими порциями при перемешивании содержимого стеклянной палочкой. После завершения процесса осушки экстракт сливают в мерный цилиндр вместимостью 25 см3, затем делят его на две приблизительно равные части (экстракт № 1 и экстракт № 2).
Экстракт № 1 заливают в измерительную кювету, которую предварительно ополаскивают этим раствором, и проводят измерение суммарной концентрации НП и Ж в соответствии с 10.3.
Экстракт № 2 пропускают через хроматографическую колонку с оксидом алюминия, подготовленную по 9.2.5 следующим образом. В хроматографическую колонку наливают 3 см3 четыреххлористого углерода для смачивания. Как только четыреххлористый углерод впитается в оксид алюминия, пропускают экстракт № 2 через хромотографическую колонку. Первые 3 см3 элюата отбрасывают, а оставшуюся часть элюата собирают в мерный цилиндр вместимостью 10 - 25 см3.
Проведение измерений осуществляют в соответствии с руководством по эксплуатации концентратомера.
Анализируемый раствор (экстракт № 1 или элюат, полученные по 10.2) заливают в кювету и устанавливают в прибор. Измеряют концентрацию, считывая показания прибора.
В случае если концентрация анализируемого раствора превышает верхнюю границу диапазона измерений прибора, то разбавляют раствор четыреххлористым углеродом, подготовленным по 9.2.1. Затем раствор заливают в кювету, которую предварительно ополаскивают этим раствором, устанавливают в прибор и производят измерение.
11. ОПРЕДЕЛЕНИЕ НП И Ж В ХОЛОСТОЙ ПРОБЕ
Определение массовой концентрации НП и Ж в холостой пробе выполняют одновременно с анализом серии проб. Для этого берут 20 см3 четыреххлористого углерода и обрабатывают, как описано в 10.2.
Результаты анализа холостой пробы учитывают при расчете концентрации НП и Ж в пробе.
Анализ холостой пробы проводят также при использовании новой партии реактивов.
12. ОБРАБОТКА РЕЗУЛЬТАТОВ ИЗМЕРЕНИЯ
12.1. Массовую концентрацию Х1, мг/дм3, экстрагированных веществ (НП и Ж) в анализируемой пробе воды рассчитывают по формуле
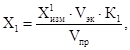 (1)
(1)
где ![]() - суммарная
массовая концентрация НП и Ж, измеренная на приборе в экстракте № 1 за вычетом
результата измерения соответствующей холостой пробы, мг/дм3;
- суммарная
массовая концентрация НП и Ж, измеренная на приборе в экстракте № 1 за вычетом
результата измерения соответствующей холостой пробы, мг/дм3;
Vэк - объем четыреххлористого углерода, использованного для проведения экстракции (Vэк = 20 см3);
К1 - коэффициент разбавления, т.е. соотношение объемов мерной колбы и аликвоты экстракта № 1 (учитывается при его разбавлении по 10.3);
Vnp - объем анализируемой пробы воды, см3.
12.2. Массовую концентрацию Х2 (мг/дм3) НП в анализируемой пробе воды рассчитывают по формуле
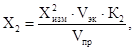 (2)
(2)
где
![]() -
массовая концентрация НП, измеренная на приборе в элюате за вычетом результата
измерения соответствующей холостой пробы, мг/дм3;
-
массовая концентрация НП, измеренная на приборе в элюате за вычетом результата
измерения соответствующей холостой пробы, мг/дм3;
Vэк - объем четыреххлористого углерода, использованного для проведения экстракции (Vэк = 20 см3);
К2 - коэффициент разбавления, т.е. соотношение объемов мерной колбы и аликвоты элюата (учитывается при его разбавлении по 10.3);
Vnp - объем анализируемой пробы воды, см3.
12.3. Массовую концентрацию X, мг/дм3, Ж в анализируемой пробе воды рассчитывают по формуле
Х = Х1 - Х2, (3)
где Х1 - суммарная массовая концентрация НП и Ж, рассчитанная по формуле (1), мг/дм3;
Х2 - массовая концентрация НП, рассчитанная по формуле (2), мг/дм3.
12.4. За результат измерения массовой концентрации НП, Ж принимают среднее арифметическое значение X (мг/дм3) двух результатов параллельных определений (Х1, Х2), расхождение между которыми для диапазона не должно превышать предела повторяемости - r. Значения предела повторяемости (r) для двух результатов параллельных определений приведены в таблице 3.
При превышении предела повторяемости (r) необходимо дополнительно получить еще два результата параллельных определений. Если при этом размах (Хmax - Хmin) четырех результатов параллельных определений равен или меньше критического диапазона CR0,95(4), то в качестве окончательного результата принимают среднее арифметическое значение четырех результатов параллельных определений. Значения CR0,95 приведены в таблице 3.
Если размах (Хmax - Хmin) > CR0,95(4), то в качестве окончательного результата измерения может быть принята медиана результатов четырех определений. Кроме того, целесообразно выяснить причины появления неприемлемых результатов параллельных определений.
Таблица 3 - Диапазон измерений, значения предела повторяемости и критического диапазона при доверительной вероятности P = 0,95
|
Предел повторяемости (относительное значение допускаемого расхождения между двумя результатами параллельных определений), r, % |
Относительное значение критического диапазона четырех результатов параллельных определений, CR0,95(4) % |
|
|
нефтепродукты |
||
|
От 0,04 до 0,25 вкл. |
28 |
36 |
|
Свыше 0,25 до 0,5 вкл. |
25 |
33 |
|
Свыше 0,5 до 5,0 вкл. |
22 |
29 |
|
жиры |
||
|
От 0,1 до 0,5 вкл. |
22 |
29 |
|
Свыше 0,5 до 10,0 вкл. |
14 |
18 |
13. ОФОРМЛЕНИЕ РЕЗУЛЬТАТОВ ИЗМЕРЕНИЙ
Результат измерения (![]() ), мг/дм3, в документах,
предусматривающих его использование, может быть представлен в виде
), мг/дм3, в документах,
предусматривающих его использование, может быть представлен в виде
![]() P = 0,95, (4)
P = 0,95, (4)
где
![]() (
(![]() - массовая
концентрация НП или Ж в пробе), значения δ приведены в таблице 1.
- массовая
концентрация НП или Ж в пробе), значения δ приведены в таблице 1.
Допустимо результат измерения в документах, выдаваемых лабораторией, представлять в виде
![]() P = 0,95 при
условии Dл < D, (5)
P = 0,95 при
условии Dл < D, (5)
где
![]() -
результат измерения, полученный в соответствии с прописью методики;
-
результат измерения, полученный в соответствии с прописью методики;
±Dл - значения характеристики погрешности результатов измерений, установленное при реализации методики в лаборатории и обеспечиваемое контролем стабильности результатов измерений.
Примечание - Допустимо характеристику погрешности результатов измерений при внедрении методики в лаборатории устанавливать на основе выражения: Dл = 0,84 · D, с последующим уточнением по мере накопления информации в процессе контроля стабильности результатов измерений.
14. ПРОВЕРКА ПРИЕМЛЕМОСТИ РЕЗУЛЬТАТОВ, ПОЛУЧАЕМЫХ В УСЛОВИЯХ ВОСПРОИЗВОДИМОСТИ
14.1. Расхождение между результатами измерений, полученными в двух лабораториях, не должно превышать предела воспроизводимости. При выполнении этого условия приемлемы оба результата измерений, и в качестве окончательного может быть использовано их общее среднее значение. Значения предела воспроизводимости (R) приведены в таблице 4.
14.2. При превышении предела воспроизводимости могут быть использованы методы проверки приемлемости результатов измерений согласно раздела 5 ГОСТ Р ИСО 5725-6.
Таблица 4 - Диапазон измерений, значения предела воспроизводимости при доверительной вероятности P = 0,95
|
Предел воспроизводимости (относительное значение допускаемого расхождения между двумя результатами измерений, полученными в разных лабораториях), R |
|
|
нефтепродукты |
|
|
От 0,04 до 0,25 вкл. |
50 |
|
Свыше 0,25 до 0,5 вкл. |
39 |
|
Свыше 0,5 до 5,0 вкл. |
34 |
|
жиры |
|
|
От 0,1 до 0,5 вкл. |
36 |
|
Свыше 0,5 до 10,0 вкл. |
34 |
15. КОНТРОЛЬ КАЧЕСТВА РЕЗУЛЬТАТОВ ИЗМЕРЕНИЙ ПРИ РЕАЛИЗАЦИИ МЕТОДИКИ В ЛАБОРАТОРИИ
15.1. Контроль качества результатов измерений при реализации методики в лаборатории предусматривает:
- контроль исполнителем процедуры выполнения измерений (на основе погрешности при реализации отдельно взятой контрольной процедуры);
- контроль стабильности результатов измерений (на основе контроля стабильности среднеквадратичного отклонения внутрилабораторной прецизионности, погрешности).
15.2. Алгоритм контроля процедуры выполнения измерений с использованием метода добавок
15.2.1. Контроль исполнителем процедуры выполнения измерений проводят путем сравнения результата отдельно взятой контрольной процедуры Кк с нормативом контроля точности К.
15.2.2. Результат контрольной процедуры Кк рассчитывают по формуле
![]() (6)
(6)
где
![]() -
результат контрольного измерения содержания определяемого компонента (НП или Ж) в пробе с добавкой - среднее арифметическое
двух результатов параллельных определений, расхождение между которыми не
превышает предела повторяемости r;
-
результат контрольного измерения содержания определяемого компонента (НП или Ж) в пробе с добавкой - среднее арифметическое
двух результатов параллельных определений, расхождение между которыми не
превышает предела повторяемости r;
![]() - результат
контрольного измерения содержания определяемого компонента в рабочей пробе (НП
или Ж) - среднее арифметическое двух результатов параллельных определений,
расхождение между которыми не превышает предела повторяемости r. Значение r приведено в
таблице 3;
- результат
контрольного измерения содержания определяемого компонента в рабочей пробе (НП
или Ж) - среднее арифметическое двух результатов параллельных определений,
расхождение между которыми не превышает предела повторяемости r. Значение r приведено в
таблице 3;
Сд - величина добавки (НП и Ж).
15.2.2. Норматив контроля К рассчитывают по формуле
![]() (7)
(7)
где
![]() (
(![]() ) - значение
характеристики погрешности результатов измерений, установленное в лаборатории
при реализации методики, соответствующее содержанию компонента НП или Ж в пробе
с добавкой (рабочей пробе соответственно).
) - значение
характеристики погрешности результатов измерений, установленное в лаборатории
при реализации методики, соответствующее содержанию компонента НП или Ж в пробе
с добавкой (рабочей пробе соответственно).
Значение характеристики погрешности рассчитывают по формуле
![]() (8)
(8)
где δл - относительное значение характеристики погрешности результатов измерений, установленное в лаборатории при реализации методики установлены в лаборатории.
15.2.3. Качество контрольной процедуры признают удовлетворительным при выполнении условия
Кк £ К. (9)
При невыполнении условия (9) эксперимент повторяют. При повторном невыполнении условия (9) выясняют причины, приводящие к неудовлетворительным результатам.
15.2.4. Периодичность контроля исполнителем процедуры выполнения измерений, а также реализуемые процедуры контроля стабильности результатов выполняемых измерений регламентируют в Руководстве по качеству лаборатории.
ФЕДЕРАЛЬНОЕ АГЕНТСТВО ПО ТЕХНИЧЕСКОМУ РЕГУЛИРОВАНИЮ И МЕТРОЛОГИИ Государственный научный метрологический центр ФГУП «Уральский научно-исследовательский институт метрологии» СВИДЕТЕЛЬСТВО № 223.1.01.05.59/2009 Методика выполнения измерений массовых концентраций нефтепродуктов и наименование измеряемой величины: объекта жиров (при их совместном присутствии) в питьевых, природных и очищенных сточных водах ___________________________________________________________ и метоlа измерений _________________________________________________________________________ методом ИК-спектрофотометрии на концентратомере КН-2м, ________________ разработанная ООО «Производственно-экологическое предприятие «СИБЭКОПРИБОР» _________________________________________________________________________ наименование организации (предприятия), разработавшей МВИ аттестована в соответствии с ГОСТ Р 8.563, ________________________________ Аттестация осуществлена по результатам метрологической экспертизы материалов ______________________________________________________________ вид: работ метрологическая экспертиза материалов по разработке МВИ. по разработке методики выполнения измерений ____________________________ теоретическое или экспериментальное исследование МВИ, другие виды работ В результате аттестации установлено, что МВИ соответствует предъявляемым к ней метрологическим требованиям и обладает следующими основными метрологическими характеристиками, приведенными в приложении. Приложение: метрологические характеристики МВИ на 1 листе Зам. директора по научной работе С.В. Медведевских Зав. лабораторией Г.И. Терентьев Дата выдачи: 10.06.2009 г. Срок действия: 10.06.2014 г.
|
Приложение к свидетельству об аттестации №
223.1.01.05.59 /2009
методики выполнения измерений массовых концентраций нефтепродуктов и жиров (при
их совмеcтном присутствии) в питьевых, природных и
очищенных сточных водах методом ИК-спектрофотометрии
1. Диапазон измерений, значения показателей точности1, повторяемости и воспроизводимости
___________
1 соответствует относительной расширенной неопределенности с коэффициентом охвата k = 2
|
Диапазон измерений, мг/дм3 |
Показатель повторяемости (относительное значение среднеквадратического отклонения повторяемости). sr,% |
Показатель воспроизводимости (относительное значение среднеквадратического отклонения воспроизводимости при n = 1), sR, % |
Показатель воспроизводимости (относительное значение
среднеквадратического отклонения воспроизводимости при n = 2), |
Показатель точности (границы относительной погрешности при вероятности Р = 0 95 и n =1, ± δ, % |
Показатель точности (границы относительной
погрешности при вероятности Р = 0,95 и n = 2), ± |
|
Нефтепродукты (питьевые, природные и очищенные сточные воды) |
|||||
|
от 0,04 до 0,25 включ. |
10 |
18 |
16 |
36 |
32 |
|
св. 0,25 « 0,5 « |
9 |
14 |
12 |
28 |
24 |
|
« 0,5 « 5 « |
8 |
12 |
10 |
24 |
20 |
|
Жиры (питьевые воды) |
|||||
|
от 0,1 до 0,5 включ. |
8 |
12,5 |
11 |
25 |
22 |
|
св. 0,5 « 10 « |
5 |
10 |
9 |
20 |
18 |
|
Жиры (природиые и очищенные сточные воды) |
|||||
|
от 0,1 до 0,5 включ. |
8 |
13 |
12 |
32 |
30 |
|
св. 0,5 « 10 « |
5 |
12 |
11 |
26 |
24 |
|
Примечание - n - количество результатов параллельных определений, необходимых для получения окончательного результата измерений |
|||||
2. Диапазон измерений, значения пределов повторяемости, воспроизводимости и критической разности при вероятности Р = 0,95
|
Диапазон измерений, мг/дм3 |
Предел повторяемости (относительное значение допускаемого расхождения между двумя результатами параллельных определений), r, % |
Предел воспроизводимости (относительное значение допускаемого расхождения между двумя единичными результатами измерений, полученными в разных лабораториях при n1 = n2 = 1), R, % |
Критическая разность (относительное значение допускаемого расхождения между двумя средними арифметическими результатами измерениями, полученными в разных лабораториях при n1 = n2 = 2), CD0,95, % |
|
Нефтепродукты (питьевые, природные и очищенные сточные воды) |
|||
|
От 0,04 до 0,25 включ. |
28 |
50 |
45 |
|
св. 0,25 « 0,5 « |
25 |
39 |
34 |
|
« 0,5 « 5 « |
22 |
34 |
28 |
|
Жиры (питьевые воды) |
|||
|
от 0,1 до 0,5 включ. |
22 |
35 |
31 |
|
св. 0,5 « 10 « |
14 |
28 |
25 |
|
Жиры (природные и очищенные сточные воды) |
|||
|
от 0,1 до 0,5 включ. |
22 |
36 |
34 |
|
св. 0,5 « 10 « |
14 |
34 |
31 |
|
Примечание - n1 - количество результатов параллельных определений, полученных в первой лаборатории; n2 - количество результатов параллельных - определений, полученных во второй лаборатории. |
|||
3. Контроль стабильности результатов измерений, получаемых в условиях повторяемости и промежуточной (внутрилабораторной) прецизионности, организуют и проводят в соответствии с ГОСТ Р ИСО 5725-6-2002. Периодичность получения результатов контрольных процедур и формы их регистрации приводят в документах лаборатории, устанавливающих порядок и содержание работ по организации методов контроля стабильности результатов измерений в пределах лаборатории.
|
Старший научный сотрудник |
О.В. Кочергина |
СОДЕРЖАНИЕ