ГОСУДАРСТВЕННЫЙ КОМИТЕТ СССР
ПО ГИДРОМЕТЕОРОЛОГИИ И КОНТРОЛЮ ПРИРОДНОЙ СРЕДЫ
ИНСТИТУТ ЭКСПЕРИМЕНТАЛЬНОЙ МЕТЕОРОЛОГИИ
ВРЕМЕННЫЕ
МЕТОДИЧЕСКИЕ РЕКОМЕНДАЦИИ
ПО КОНТРОЛЮ ЗАГРЯЗНЕНИЯ ПОЧВ
Часть II
Под
редакцией
канд. физ.-мат. наук С. Г. МАЛАХОВА
МОСКВА. МОСКОВСКОЕ ОТДЕЛЕНИЕ ГИДРОМЕТЕОИЗДАТА - 1984
Вторая часть "Временных методических рекомендаций по контролю загрязнения почв включает в себя описание контроля загрязнения почв нефтепродуктами, а также комплекс методов, позволяющих оценивать изменения агрохимических и биологических свойств почвы под влиянием загрязняющих веществ. К последним относятся методы определения ферментативной активности, интенсивности дыхания почвы, содержания в почве углерода, фосфора, азота и сульфатов.
Рекомендации предназначены для лабораторий контроля загрязнения почв Госкомгидромета СССР как части Общегосударственной службы наблюдения и контроля загрязнения почв. Они будут полезны работникам лабораторий других ведомств, занимающихся определением уровней загрязнения почв, а также научным работникам и специалистам, работающим над проблемой охраны почв от загрязнений или интересующихся ею.
ПРЕДИСЛОВИЕ
Настоящие рекомендации являются продолжением (второй частью) "Временных методических рекомендаций по контролю загрязнения почв", изданных в 1983 г. Гидрометеоиздатом.
В первую часть Рекомендаций вошли методики отбора проб почвы, методики определения в этих пробах хлорорганических и фосфорорганических пестицидов, гербицидов (2,4-Д-симтриазинов), валовых количеств металлов, форм соединений металлов, фторидов, методика определения рН почвы и выпадений. В первой части кратко описана методика определения выпадений металлов из атмосферы. Кроме того, отдельно даны рекомендации по метрологическому обеспечению проводимых измерений (по методике внутреннего и внешнего контроля достоверности результатов измерений).
Настоящая (вторая) часть Рекомендаций включает методику определения содержания в почвах нефтепродуктов. Основное же внимание уделено методам оценок последствий загрязнения почв - методам оценок изменения основных агрохимических и биологических свойств почвы под влиянием антропогенного загрязнения. Во второй части приведены методики определения содержания в почвах углерода, подвижного фосфора, аммиачного азота, нитратного азота, обменных сульфатов, методики измерения ферментативной активности почв.
В составлении отдельных разделов второй части методических рекомендаций принимали участие: сотрудник МГУ им. М.В. Ломоносова канд. геогр. наук Ю.И. Пиковский, сотрудники ИЭМ: канд. хим. наук Л.С. Эрнестова (раздел IV), канд. биол. наук Э.И. Гапонюк (разделы V и VI), м.н.с. Н.П. Кремленкова (раздел V и VI).
Общая редакция осуществлена канд. физ.-мат. наук С.Г. Малаховым.
РАЗДЕЛ IV. КОНТРОЛЬ ЗАГРЯЗНЕНИЯ ПОЧВ НЕФТЕПРОДУКТАМИ
IV.1. Введение
Загрязнением почв нефтью (Н) и нефтепродуктами (НП) считается увеличение концентраций этих веществ до такого уровня, при котором:
нарушается экологическое равновесие в почвенной системе;
происходит изменение морфологических, физико-химических и химических характеристик почвенных горизонтов;
изменяются водно-физические свойства почв;
нарушается соотношение между отдельными фракциями органического вещества почвы, в частности между липидной и гумусовой составляющими;
создается опасность вымывания из почвы Н и НП и вторичного загрязнения грунтовых и поверхностных вод.
Уровень допустимой концентрации Н и НП в почвах, при котором не наблюдается перечисленных выше явлений, не везде одинаков. Он будет различаться в зависимости от:
почвенно-климатической зоны;
типа почвы;
состава Н и НП, попавших в почву.
В среднем нижний предел концентраций Н и НП в загрязненной почве изменяется от 0,1 до 1,0 г/кг. Критерием также может служить концентрация выше 0,05 мг/л Н и НП в воде, профильтрованной через загрязненную почву.
Контроль за загрязнением почв нефтью и нефтепродуктами осуществляется вблизи наиболее вероятных мест импактного загрязнения:
нефтепромыслов, нефтепроводов, нефтеперерабатывающих заводов, нефтехранилищ. Основные задачи контроля состоят в следующем:
определение источника и центра разлива Н и НП;
определение потока нефти по площади и по глубине почвенного профиля;
определение направления движения потока и возможного ареала дальнейшего загрязнения;
идентификация продуктов загрязнения;
установление характера сопутствующего загрязнения почв (минеральными солями, токсичными металлами, канцерогенными веществами);
установление степени и характера трансформации почв и растительности, загрязненности вод;
определение возможности самоочищения почв и эффективности мероприятий по ликвидации последствий загрязнения;
оценка ущерба, нанесенного природе и сельскому хозяйству. Основной метод контроля - изучение морфологии почвенного профиля, определение содержания Н и НП в образцах почв и грунтовых вод.
В настоящем разделе дана краткая характеристика главных потенциальных источников загрязнения почв Н и НП, приводятся методика отбора проб, методика диагностики нефтяных загрязнений в почвах и определения концентраций нефти и нефтепродуктов.
IV.2. Характеристика нефти и продуктов ее переработки
Нефть - маслянистая жидкость, представляющая собой сложный природный раствор органических соединений, в основном углеводородов. В углеводородах растворены высокомолекулярные смолисто-асфальтеновые вещества, а также низкомолекулярные кислород-, азот- и серусодержащие органические соединения. Кроме того, в нефти растворены и некоторые неорганические вещества: вода, соли, сероводород, соединения металлов и других элементов.
Нефть в природе довольно разнообразна. По внешнему виду она различается по цвету (от почти бесцветной до темно-коричневой) и вязкости (от весьма подвижной до густой малоподвижной). Соотношение компонентов, входящих в состав нефти, определяет ее тип, физические свойства, состав. Изменение состава и свойств нефти отражается прежде всего на удельном весе, который колеблется от 0,80 до 0,95. Нефть с большим или меньшим удельным весом встречается редко.
В составе нефти различают следующие классы углеводородов:
алифатические (метановые);
циклические насыщенные (нафтеновые);
циклические ненасыщенные (ароматические).
Имеются также смешанные (гибридные) углеводороды: метано-нафтеновые, нафтеново-ароматические.
Среди метановых углеводородов в нефти имеются газообразные, жидкие и твердые. Газообразные (метан, этан, бутан и др.) растворены в жидких углеводородах и выделяются при изменении давления. Твердые высокомолекулярные углеводороды (парафины) также находятся в растворенном состоянии. Их попадание в почву особенно опасно, так как, имея низкую температуру застывания, парафины прочно закупоривают все каналы, по которым происходит обмен веществ между почвой и растением, почвой и атмосферой.
Нефть с преобладанием метановых углеводородов относится к метановому типу. Среди ее разновидностей выделяется высокопарафинистая нефть (содержание парафина более 6 %), парафинистая (1,5-6,0 %) и малопарафинистая (менее 1,5 %).
Нафтеновые углеводороды присутствуют во всех типах нефти, но нефть с преобладанием этого класса углеводородов встречается редко. Среди ароматических углеводородов преобладают низкомолекулярные структуры (бензол, толуол, ксилол, нафталины). В подчиненном количестве имеются гомологи 3-6-кольчатых углеводородов (полициклические ароматические углеводороды - ПАУ). В некоторых разновидностях нефти ПАУ содержат значительное количество 3,4-бенз(а)пирена и других канцерогенных углеводородов.
Высокомолекулярные ароматические структуры, содержащие также кислород, серу, азот, представляют смолы и асфальтены. Смолы - вязкие вещества, асфальтены - твердые. Те и другие растворены в жидких углеводородах. Высокое содержание смол и асфальтенов в нефти определяет увеличение ее удельного веса и вязкости. Такие нефти малоподвижны, но могут создать устойчивый очаг загрязнения в почве.
Смолистые нефти не содержат, как правило, твердых парафинов, а высокопарафинистые нефти - заметного количества смолисто-асфальтеновых веществ. Нафтеновые нефти содержат минимальное количество тех и других.
Существенное значение имеет содержание серы в нефти. Кроме элементной серы, в нефти присутствуют некоторые специфические сернистые соединения (меркаптаны, сульфиды, тиофаны), дающие специфический запах. Присутствие сернистых соединений увеличивает токсичность нефти.
По содержанию серы нефть бывает малосернистая (менее 0,5 %), сернистая (0,5-2,0%), высокосернистая (более 2,0%).
Нефть разделяется на фракции по температуре кипения смесей различных углеводородов. Углеводороды, вскипающие до 200 °С, относятся к бензиновой, вскипающие в интервале 200-300 °С, - керосиновой, от 300 до 400 °С - газоилевой фракциям.
Нефть, богатая бензиновой фракцией, быстрее испаряется, и ее воздействие на природную среду относительно кратковременно. Углеводороды, вскипающие при высоких температурах, довольно устойчивы и очищение от них компонентов природной среды проходит с трудом.
В табл. IV.2.1 отмечены некоторые свойства разных типов нефти. Тип нефти зависит от многих условий, связанных с формированием и существованием ее скоплений. В одном и том же районе можно встретить разные типы нефти. Часто наблюдается определенная зональность по площади и по толще нефтеносных пород в изменении состава нефти.
Из нефти получают несколько тысяч различных продуктов, которые делятся на следующие основные группы:
топлива (бензины, лигроины, керосины, реактивные, дизельные, котельные, газотурбинные топлива);
нефтяные масла;
парафины, церуины, вазелины;
нефтяные битумы;
осветительные керосины;
растворители;
прочие нефтепродукты (кокс, сажа, смазки, органические кислоты и др.).
Таблица IV.2.1
Классификация нефти по углеводородному составу и некоторым физико-химическим свойствам (по А.Ф. Добрянскому)
|
Классы нефти |
||||||
|
Ароматические, ароматическо-нафтеновые |
нафтеновые, нафтеново-ароматические |
метано-нафтеновые |
метановые |
|||
|
Удельный вес при 20 °С |
0,9-0,95 |
0,87-0,91 |
0,83-0,87 |
0,80-0,83 |
||
|
Вязкость при. 50 °С |
2-10 |
1-3 |
1-2 |
1-1,5 |
||
|
Групповой состав углеводородов: |
|
метановые |
0-10 |
5-20 |
20-40 |
40-55 |
|
нафтеновые |
46-60 |
50-60 |
45-60 |
35-45 |
||
|
ароматические |
35-55 |
20-40 |
10-25 |
5-10 |
||
|
Выход бензиновых фракций, 200 °С |
5-15 |
10-25 |
25-35 |
30-45 |
||
Нефтепродукты состоят из тех же компонентов сырой нефти, отделенных друг от друга и полученных из них путем термокаталитических химических реакций. Топлива, например, - это жидкие углеводородные продукты. Они различаются по температуре кипения:
начало кипения - 200 °С - бензины (авиационные, автомобильные топлива);
200 - 300 °С - керосины, лигроины (тракторные, реактивные топлива);
>300 °С - мазуты, газойли (котельные, газотурбинные топлива).
Нефтяные битумы, наоборот, - это твердые продукты, лишенные углеводородной составляющей. Они применяются в строительстве, производстве изоляционных материалов, резины и др.
Парафины, церезины - это твердые углеводороды, находящие применение в нефтехимии, пищевой промышленности, медицине.
Масла - вязкие продукты, состоящие из высокомолекулярных углеводородов. Применяются для смазок, гидропередач, изоляций и других целей.
Природную среду загрязняют в широких масштабах, главным образом, сырая нефть, топлива, масла, нефтяные битумы, сажа. Наиболее распространены первые две группы веществ, самые подвижные из всех. Через атмосферу широко распространяется сажа.
Токсичность разных типов нефти и нефтепродуктов не одинакова.
Легкие фракции нефти и легкие нефтепродукты (бензины, керосины) обладают наиболее сильным токсическим действием на живые организмы. Но влияние этих продуктов происходит непродолжительное время вследствие быстрого испарения, биодеградации и рассеяния.
Тяжелые фракции нефти и тяжелые нефтепродукты сильного токсического действия на организм не оказывав, но они значительно ухудшают свойства почв, затрудняют газо- и водообмен в почвах, затрудняют дыхание и питание растений. Эти компоненты очень устойчивы и могут сохраняться в почвах в течение длительного времени (годы, десятки лет).
Важное свойство нефти и нефтепродуктов - растворимость. Сами нефти и нефтепродукты хорошо растворяют различные неорганические и органические вещества, среди которых могут находиться весьма токсичные соединения. Например, из неорганических веществ - это сера, сернистые соединения и другие. Из органических - канцерогенные углеводороды и многие другие токсичные и ядовитые вещества.
Нефть и нефтепродукты при комнатных и более низких температурах в воде практически не растворяются. В среднем их растворимость составляет сотые доли процента. Но уже этого достаточно, чтобы резко ухудшить качество воды. Обычно нефтяные компоненты образуют с водой эмульсию, которую трудно разрушить. Чаще всего нефть плавает на поверхности воды в виде пленки, обволакивает взвешенные частицы и оседает с ними на дно.
Из отдельных классов углеводородов лучше в воде растворяются ароматические, хуже - метановые. Растворимость углеводородов в воде снижается от низкомолекулярных к высокомолекулярным соединениям.
Нефть и нефтепродукты хорошо растворяются в малополярных органических растворителях. Практически все нефтяные компоненты полностью растворимы в бензоле, хлороформе, диэтиловом эфире, сероуглероде, четыреххлористом углероде. Все эти вещества весьма токсичные. Несколько менее других опасен хлороформ.
Неполярные органические растворители - петролейный эфир, гексан - растворяют всю углеводородную часть нефти, но не растворяют входящие в ее состав асфальтены и высокомолекулярные смолы. Поскольку содержание асфальтенов в неизменной нефти обычно мало (1-2 %), то этими растворителями часто пользуются для диагностики загрязнений: они не растворяют полярные органические соединения, не имеющие отношения к нефти и нефтепродуктам.
Нефти, нефтепродукты и другие близкие им по составу соединения, находящиеся в природной среде (почвах, грунтах, горных породах), имеют собирательное название "битуминозные вещества". Сумму битуминозных веществ, извлеченных из этих природных объектов органическими растворителями, называют битумоидами. Растворы битумоидов обладают способностью люминесцировать в ультрафиолетовых лучах. Это свойство можно использовать для диагностики загрязнений в почвах, изучения качественных и количественных характеристик загрязняющих веществ [3].
IV.3. Возможные источники и очаги загрязнения почв нефтью и нефтепродуктами
Нефть и нефтепродукты рассеиваются в окружающей природной среде повсеместно, так как в современном мире нет такой области хозяйственной деятельности человека, где бы они не использовались. В области, свободной от хозяйственной деятельности человека (заповедники, труднодоступные территории), углеводороды транспортируются с воздушными и водными потоками. Глобальное или региональное рассеяние углеводородов происходит, как правило, из суммы источников, находящихся иногда на значительных расстояниях и мало связанных между собой.
Актуальное практическое значение представляют собой импактные загрязнения природной среды нефтью и нефтепродуктами. Такие загрязнения, имеющие, как правило, конкретный источник, создают значительную единовременную нагрузку на почву, воду, биологические объекты, нанося порой большой ущерб народному хозяйству и природе. Импактные загрязнения - основной объект контроля в настоящее время.
Главные потенциальные источники загрязнения природной среды нефтью и нефтепродуктами - это нефтепромыслы, нефтепроводы, нефтеперерабатывающие предприятия, нефтехранилища, наземный и водный транспорт, перевозящий нефтепродукты. Характеристика этих источников приведена в табл. IV.3.1.
Таблица IV.3.1
Главные потенциальные источники загрязнения природной среды нефтью и нефтепродуктами
|
Источник загрязнения |
Основные причины загрязнений |
Вещества, загрязняющие природную среду |
|
|
Нефтепромысел |
Скважины |
Стравливание во время ремонта, нарушение герметичности, арматуры, аварийные выбросы |
Сырая нефть, товарная нефть, минерализованные воды |
|
Трубопроводы |
Коррозия и механические повреждения труб |
NaCl, CaSO4 и др. |
|
|
Сборные пункты, нефтехранилища |
Испарение углеводородов в атмосферу, утечки в результате нарушения герметичности емкостей |
Конденсаты |
|
|
Пункты первичной подготовки нефти Факелы |
То же, что на сборных пунктах и трубопроводах; сброс сточных вод |
Конденсаты, сажа, канцерогенные углеводороды |
|
|
Неполное сгорание нефтепродуктов, конденсация стравленных в воздухе углеводородов |
Конденсаты, сажа, канцерогенные углеводороды, сернистые соединения |
||
|
Нефтепроводы |
Нефтепроводы, нефтепродуктопроводы |
Механические повреждения труб, коррозия |
Товарная нефть (обезвоженная и обессоленная), жидкие нефтепродукты |
|
Нефтеперерабатывающие заводы, нефтехранилища |
Очистные сооружения, канализация |
Аварии, разгерметизация соединений трубопроводов, испарение нефтепродуктов в атмосферу |
Сточные воды с нефтью и нефтепродуктами (от 100 до 15000 мг/л) |
|
Резервуары для хранения нефтепродуктов |
Выбросы в атмосферу через клапаны при избыточном давлении паров, нарушение герметичности резервуаров |
Легкие углеводороды, мазуты, дизельные и другие топлива |
|
|
Технологические установки |
Выбросы через предохранительные клапаны |
Углеводороды, сероводород |
|
|
Факельные системы |
Неполное сгорание углеводородов, сероводорода, отсутствие пламени на факеле |
Углеводороды, сероводород, окислы серы, углерода, фенолы, бензол, бенз(а)пирен |
Наиболее распространенный и менее всего управляемый источник - нефтепроводы, по которым перекачивается сырая и товарная нефть, а также различные жидкие нефтепродукты. Нефтепроводы густой сетью располагаются в нефтедобывающих районах, их нитки протягиваются через всю страну, пересекая реки, каналы, горные хребты. Аварии нефтепроводов часто случаются вблизи рек, которыми нефть разносится на большие расстояния.
На территориях нефтепромыслов главными источниками загрязнения являются эксплуатационные и разведочные скважины, из которых происходят аварийные выбросы. На отдельных промыслах число таких скважин достигает нескольких сот. На нефтепромыслах имеются и другие источники загрязнения: трубопроводы, сборные пункты, хранилища, пункты подготовки нефти. В зависимости от положения нефтепромысла в ландшафтно-геохимической системе потоки нефти и нефтяных вод могут захватывать и смежные территории.
Нефтеперерабатывающие предприятия и заводы (НПЗ) и нефтехранилища - локальные источники загрязнения. Они загрязняют среду главным образом через атмосферу и сточные воды. Единовременные выбросы на почву при этом относительно невелики, но их постоянное действие создает вокруг значительный ареал устойчивого загрязнения. Например, на НПЗ производительностью 12 млн. т нефти в год только через предохранительные клапаны на технологических установках выбрасывается в атмосферу около 100 т углеводородов в сутки [5].
IV.4. Обследование мест импактного загрязнения почв нефтью и нефтепродуктами
Потоки нефти и нефтепродуктов в почвах могут быть видимыми и скрытыми (внутрипочвенными). Видимые потоки оконтуриваются визуально. В этих случаях источник загрязнения определяется без затруднений.
Скрытые потоки возникают чаще всего в результате аварий трубопроводов, проходящих на некоторой глубине от поверхности земли. Появление скрытых потоков нефти фиксируется по резкому увеличению содержания нефтепродуктов в грунтовых водах, находящихся поблизости от источника загрязнения, поверхностных водах (реках, ручьях, каналах, озерах, прудах). Внутрипочвенные потоки проявляют себя высачиванием нефти на склонах, стенках канав, кюветов. Скрытое загрязнение может быть зафиксировано по изменению растительного покрова: пожелтению травянистой растительности, засыханию деревьев и кустарников.
Для оконтуривания нефтяного потока по площади и по вертикали и для определения места разлива необходимо определить ландшафтно-геохимическую позицию исследуемого участка [9]:
1) тип элементарного ландшафта (автономный - на плоской возвышенности, трансэлювиальный - на склоне; элювиально-аккумулятивный - в небольших местных понижениях рельефа; транссупераквальный - подножие склона, поймы рек; трансаквальный - реки и другие водотоки);
2) типы геохимических сопряжений в местных ландшафтах, которые определяют характер перемещения вещества: соотношение бокового и вертикального стоков; формы миграции, характер геохимических и физических барьеров, задерживающих нефть на пути движения потока.
При определении типов сопряжении важное значение имеют:
а) глубина просачивания атмосферных вод; б) глубина залегания грунтовых вод [1].
Исходя из данных, перечисленных в пунктах I, II закладывается серия почвенных разрезов (или ручных скважин). Количество разрезов зависит от сложности ландшафтной геохимической обстановки и нефтяного потока.
Почвенные разрезы (скважины) объединяются в систему профилей, протягивающихся в направлении движения поверхностного стока от места разлива до места промежуточной или конечной аккумуляции. Минимальное количество профилей - 3, минимальное количество разрезов - 12 (по 3 на каждом профиле и 3 фоновых по одному на каждый элементарный ландшафт). Если при минимальном количестве разрезов достоверно решить задачу нельзя, закладывается необходимое количество дополнительных разрезов.
Почвенные разрезы разделяются на опорные и "приколки" (опытные образцы почв). Опорные разрезы закладываются вблизи места разлива и на основных элементах ландшафтно-геохимического профиля. Цель изучения таких разрезов - определить глубину просачивания нефти, наличие внутрипочвенного потока, характер трансформации почвенного профиля.
Изучение опорного почвенного разреза проводится так же, как и при контроле загрязнения пестицидами (см. часть 1 и [9]).
Разрез закладывается приблизительно следующих размеров:
ширина короткой стенки 0,8 м, длинной стенки - 1,5 м, глубина 2,0 м (если не вскрыты на меньшей глубине грунтовые воды). Располагается разрез так, чтобы лицевая короткая стенка была освещена солнцем. Почву выбрасывают на длинные боковые стенки: верхние горизонты - в одну сторону, нижние - в другую. На лицевой стенке производят отбор проб и по ней - описание почвы. Стенка зачищается, вдоль нее спускается сантиметр, по которому отмечаются глубины взятия проб и границы почвенных горизонтов. Отбор проб начинают с нижних горизонтов. Образец берется размером 10´10 см, а если мощность горизонта меньше, то на всю мощность.
Пробы берутся с помощью почвенного ножа. После взятия каждой пробы нож очищается от нефтепродуктов тампоном, смоченным в органическом растворителе.
Перед взятием образцов проводится описание ландшафта и почвенных горизонтов (цвет, влажность, структура, плотность, механический состав, новообразования, включения, корневая система, карбонатность).
Если выделение генетических горизонтов почв вызывает затруднение, пробы необходимо отбирать через 20 см, сопровождая их подробным описанием.
"Прикопки" для взятия почвенных образцов отрываются на глубину нижнего фронта движения нефтяного потока в почве, которую можно обычно определить по опорному разрезу.
Необходимо иметь в виду, что, если поверхность почвы или ее верхние горизонты не содержат видимых загрязнений, это не значит, что загрязнения в этом почвенном профиле отсутствуют. Нефть и нефтепродукты могут двигаться и длительное время сохраняться на глубинах 0,5-1,0 м и более под относительно плотными и мало загрязненными верхними горизонтами разреза. Поэтому изучение опорных разрезов при контроле загрязнения почв нефтью и нефтепродуктами обязательно.
Вследствие сильного варьирования состава и свойств почвы даже в пределах профиля с лицевой стороны разреза по горизонтали берется 5-8 проб для составления смешанного почвенного образца. Общий вес смешанного образца 0,6-0,8 кг [9].
IV.5. Предварительная диагностика нефтяных загрязнений в почвах
IV.5.1. Реактивы и растворы
Гексан (С6Н14) х.ч.
Хлороформ (CHCl3) х.ч.
Хроматографическая бумага марки "С", полоски 7´250 мм.
IV.5.2. Приборы и посуда
Аналитические весы ВЛР-200, АДВ-200 и т.д.
Люминесцентный осветитель любой марки (ОИ-18, ОЛД-1, ВИО-1) с фильтром УФС-6, УФС-9.
Пробирки градуированные со шлифом, ПГКШ-14, 5-25.
Цилиндры мерные объемом 50, 100 мл, ГОСТ 8682-70.
Капельница.
Сито с диаметром отверстий 0,50 мм, ТУ 46-47 885-73.
IV.5.3. Приемы предварительной диагностики
Диагностика нефтяных загрязнений в почвах проводится непосредственно в поле или в полевой лаборатории. Характер загрязнения разреза нефтью и нефтепродуктами можно предварительно определить непосредственно в разрезе. Для этого к ровной лицевой стопке разреза плотно прикладывают лист фильтровальной бумаги. В местах, где почва загрязнена нефтью и нефтепродуктами, на листе бумаги проступят масляные пятна.
Для диагностики загрязнений можно использовать метод капельного анализа по В.Н. Флоровской.
Комочки почвы просматривают в ультрафиолетовых лучах осветителя (в затемненном помещении), осторожно нанося на их поверхности капли растворителя. Наличие битуминозных компонентов дает о себе знать голубовато-белым свечением участка в месте нанесения капли. По характеру свечения можно приблизительно определить степень загрязнения [3] (табл. IV.5.1).
Таблица IV.5.1
|
Приблизительное содержание битуминозных компонентов в образце, г/кг |
||
|
пески |
суглинки |
|
|
Яркое ровное пятно |
Более 1,0 |
Более 5,0 |
|
Толстое кольцо или неровное пятно |
0,1-1,0 |
0,5-5,0 |
|
Рваное кольцо |
0,01-0,1 |
0,01-0,1 |
|
Слабое свечение отдельных точек |
Менее 0,01 |
Менее 0,01 |
IV.5.4. Люминесцентно-капиллярный безэталонный полуколичественный анализ нефтепродуктов
Полуколичественный анализ содержания нефтепродуктов [4] в почве проводят, если отсутствуют специальное оборудование и стандартные растворы и графики, характеризующие данный тип загрязнения. Методика применяется также для выбора навески почвы при более точных количественных определениях. Метод пригоден для определения следующих типов веществ: нефти, мазутов, смазочных масел, дизельных топлив.
Чувствительность метода 0,01 г/кг. Относительная ошибка до ±100 %.
IV.5.4.1. Подготовка проб и посуды к анализу. Пробу почвы (100 г) высушивают до воздушно-сухого состояния, растирают в фарфоровой ступке и просеивают через сито 0,50 мм. Затем пробу квартуют и для анализа берут навеску 1 г.
Реактивы - гексан и хлороформ - проверяют в ультрафиолетовых лучах на отсутствие люминесценции. При наличии свечения производят перегонку растворителей. Пробирки с притертыми пробками моют горячей водой с содой, высушивают и ополаскивают чистым нелюминесцирующим хлороформом. Если хлороформ в пробирке не люминесцирует, посуда готова к анализу.
IV.5.4.2. Ход определения. В стеклянную пробирку с притертой пробкой насыпают навеску почвы 1 г и заливают 10 мл чистого нелюминесцирующего гексана. После этого пробирку встряхивают несколько раз и оставляют на несколько часов (лучше на ночь). Затем пробирку просматривают в ультрафиолетовых лучах. Наличие голубого, беловато-голубого свечения свидетельствует о загрязнении почвы нефтепродуктами. 5 мл раствора переводят в стандартную стеклянную градуированную пробирку без пробки. В пробирку осторожно опускают полоску хроматической бумаги размером 7´250 мм и оставляют до полного испарения растворителя в помещении, где отсутствует колебание воздуха. После испарения растворителя полоску бумаги вынимают и просматривают в ультрафиолетовых лучах. Простым карандашом отмечают границы люминесцирующей зоны, а также фиксируют цвет люминесценции. Свечение зоны голубыми, желтыми, коричневыми тонами говорит о наличии нефтепродуктов. Измеряют ширину люминесцирующей зоны и производят приближенное определение концентраций нефтепродуктов в растворе по табл. IV.5.2. Аналогичные операции проводят с параллельной навеской в хлороформе.
Таблица IV.5.2
Расчет концентрации нефтепродуктов в почвах
|
Концентрация нефтепродуктов в растворе, г/мл |
Концентрация нефтепродуктов в почве, г/кг (навеска 1 г, раствор 10 мл) |
Ширина люминесцирующей зоны, мм |
Концентрация нефтепродуктов в растворе, г/мл |
Концентрация нефтепродуктов в почве, г/кг (навеска 1 г, раствор 10 мл) |
|
|
5 |
1×10-6 |
0,01 |
37 |
6×10-4 |
6,0 |
|
6 |
2×10-6 |
0,02 |
40 |
7×10-4 |
7,0 |
|
6,5 |
3×10-6 |
0,03 |
42 |
8×10-4 |
8,0 |
|
7 |
4×10-6 |
0,04 |
45 |
9×10-4 |
9,0 |
|
7,5 |
6×10-6 |
0,06 |
47 |
1×10-3 |
10,0 |
|
8 |
8×10-6 |
0,08 |
50 |
1,2×10-3 |
12,0 |
|
8,5 |
1×10-5 |
0,1 |
60 |
1,8×10-3 |
18,0 |
|
10,5 |
2×10-5 |
0,2 |
65 |
2,0×10-3 |
20,0 |
|
12 |
3×10-5 |
0,3 |
72 |
2,5×10-3 |
25,0 |
|
13 |
4×10-5 |
0,4 |
80 |
3,0×10-3 |
30,0 |
|
14 |
5×10-5 |
0,5 |
90 |
3,5×10-3 |
35,0 |
|
14,5 |
6×10-5 |
0,6 |
100 |
4,0×10-3 |
40,0 |
|
15,5 |
7×10-5 |
0,7 |
110 |
5,0×10-3 |
50,0 |
|
16,5 |
8×10-5 |
0,8 |
120 |
6,0×10-3 |
60,0 |
|
17 |
9×10-5 |
0,9 |
130 |
7,0×10-3 |
70,0 |
|
18 |
1×10-4 |
1,0 |
140 |
8,0×10-3 |
80,0 |
|
23 |
2×10-4 |
2,0 |
150 |
9,0×10-3 |
90,0 |
|
26 |
3×10-4 |
3,0 |
160 |
1,0×10-2 |
100,0 |
|
30 |
4×10-4 |
4,0 |
200 |
1,3×10-2 |
130,0 |
|
34 |
5×10-4 |
5,0 |
250 |
2,0×10-2 |
200,0 |
Примечание. Пользоваться таблицей можно при применении стандартных пробирок и бумаги. При использовании других типов пробирок и бумаги необходимо построить специальные графики. При анализе каждой партии проб необходимо производить контрольный холостой опыт.
IV.5.4.3. Расчет концентраций нефтепродуктов в почве. Концентрацию нефтепродуктов в пробе X определяют по формуле
![]() (IV.5.1)
(IV.5.1)
где С - концентрация нефтепродуктов, найденная по табл. IV.5.2 в соответствии с шириной люминесцирующей зоны, г/мл; V - объем растворителя, мл; m - навеска почвы, кг.
IV.5.4.4. Качественная идентификация. Качественная идентификация нефти и нефтепродуктов проводится по капиллярным вытяжкам. Прежде всего выявляют пробы, загрязненные нефтью и нефтепродуктами. Среди битумоидов, извлеченных из почв нейтральными органическими растворителями, выделяют три группы:
почвенные (фоновые);
смешанные (содержащие другие органические загрязнители);
нефтяные.
В табл. IV.5.3 отмечены некоторые диагностические признаки указанных групп битумоидов.
По капиллярным вытяжкам нефтяных битумоидов, полученных из хлороформного раствора, определяют характер нефтепродуктов и стадий их трансформации (табл. IV.5.4).
При наблюдениях за загрязнением почв нефтью и нефтепродуктами различают свежее и старое загрязнение.
Диагностика свежего загрязнения не вызывает затруднений. Капиллярные вытяжки и растворы битумоидов в этом случае обладают яркой люминесценцией.
Изменения состава и свойств нефти и нефтепродуктов в почвах во времени выражаются в постепенном уменьшении их растворимости и накоплении нерастворимых продуктов метаболизма.
В случае необходимости оценить старое загрязнение используют следующие диагностические критерии:
изменение морфологии генетических горизонтов почвенного профиля [2];
соотношение гексановых и хлороформных битумоидов.
В затруднительных случаях проводят изучение группового и фракционного состава гумуса и микробиологические исследования.
IV.6. Методика ИК спектрометрического определения нефтепродуктов в почве
IV.6.1. Основные положения
Общими принципами всех методов являются извлечение суммы неполярных и малополярных углеводородов из почвенного образца органическим растворителем и определение их концентрации в растворе с предварительной очисткой элюатов или без нее.
Таблица IV.5.3
Цвета люминесценции капиллярных вытяжек в битумоидах
|
Гексановый битумоид (ГБ) |
Хлороформный битумоид (ХБ) |
ГБ |
|
|
ХБ-ГБ |
|||
|
Почвенный (фоновый) |
Бесцветные, очень слабая люминесценция |
Розовые, розовато-серые тусклые |
< 1 |
|
Смешанный (общее региональное загрязнение) |
Голубые, голубовато-серые тусклые |
Желтые, оранжевые, беловато-желтые |
³ 1 |
|
Нефтяной |
Яркие беловато-желтые, голубые, синие |
От светло- до темно-коричневых |
> 1 |
Таблица IV.5.4
Цвет люминесценции капиллярных вытяжек нефти и различных нефтепродуктов
|
Нефтепродукты |
|
|
Синий, голубой (яркий) |
Дизельное масло, дизельное топливо, легкая нефть |
|
Голубовато-серый (тусклый) |
Нефтепродукты на начальной стадии окисления |
|
Светло-желтый, желтый |
Дизельное масло, отработанное, моторное топливо, нефть малосмолистая |
|
Светло-коричневый, коричневый |
Мазут топливный, нефть смолистая |
|
Темно-коричневый |
Битум, асфальт |
Предварительную очистку выделенных нефтепродуктов от примесей, мешающих определению соединений, проводят хроматографическими методами (в тонком слое, на бумаге, в колонке). Наиболее точными и универсальными методами являются газожидкостная хроматография или инфракрасная спектроскопия.
Предлагаемая методика определения нефтепродуктов в почве основана на экстракции нефтепродуктов из почвы четыреххлористым углеродом с одновременной очисткой элюатов на окиси алюминия в колонке. Концентрации углеводородов в пробе определяются ИК спектрометрическим методом (ИКС).
ИКС метод основан на измерении интенсивности С-Н-связей: метиленовых - СН2 и метильных - CH3 групп в области 2700-3100 см-1. Количественное определение нефтепродуктов проводят по калибровочным графикам, полученным на основании искусственной смеси углеводородов.
Для определения содержания битумонозных веществ в объектах окружающей среды используется люминесцентный метод, который базируется на пропорциональной зависимости между концентрацией люминесцирующего вещества и интенсивностью люминесценции в области малых концентраций. При этом репрезентативность метода целиком зависит от правильности подбора эталонов.
Чувствительность ИКС метода определения нефтепродуктов в почве составляет 0,02 г/кг воздушно-сухой навески.
IV.6.2. Реактивы
Алюминий окись (Аl2O3) безводный ч., МРТУ 6-09-5296-68. Перед употреблением реактив активируют прокаливанием при t=600±10 °С в течение 4 ч.
Углерод четыреххлористый осч., МРТУ 6-09-2666-65. Проверяют чистоту каждой партии на примеси органических соединений (наличие поглощения в области 2700-3100 см-1) относительно четыреххлористого углерода, перегнанного при 76,6±0,2 °С и пропущенного через колонку с активированной окисью алюминия. При наличии примесей реактив перегоняют при 76,6±0,2 °С.
Изооктан (С8Н18) х.ч., ТУ 6П-8-68.
Гексадекан (С16H34) х.ч., МРТУ 6-09-461467.
Бензол (С6Н6) х.ч., ГОСТ 5955-68.
Кальций хлористый ч., ГОСТ 4460-66.
Вата, промытая четыреххлористым углеродом.
IV.6.3. Приборы и посуда
Аналитические весы (ВЛР-200, АДВ-200 и т.д.).
Инфракрасный спектрометр типа ИКС-14, ИР-20, ИКС-2 и т.д.
Неразборные и разборные кюветы с окошками из кристаллов NaCl, стеклянные цилиндры (от спектрофотометра СФ-16 или СФ-4) и держатели для соответствующих кювет к ИК спектрометру.
Пробирки стеклянные, ГОСТ ПКСШ 14,5-20.
Хроматографическая колонка с внутренним диаметром 0,5-0,8 мм (рис. IV.6.1).
Штатив для колонок (рис. IV.6.2).
Цилиндры мерные объемом 50, 100 мл, ГОСТ 8682-70.
Стаканы мерные объемом 50, 100 мл, Н-50, Н-100 ТС, ГОСТ 10394-72.
Колбы мерные объемом 100 мл 4-100-2, ГОСТ 1770-74.
Пипетки па 1 мл 4-1-1, ГОСТ 20292-74.
Пипетки на 5 мл 7-1-5, ГОСТ 20292-74.
Сушильный шкаф Т-3.
Аппарат для перегонки растворителя.
Фарфоровая ступка с пестиком, ГОСТ 9147-73.
Мешки из плотной ткани для хранения проб.
Сито с диаметром отверстий 0,50 мм, ТУ 46-47 885-73.
Электрическая плитка ТУ 92-288-74.
IV.6.4. Подготовка образцов к анализу
Из почвенных образцов в воздушно-сухом состоянии отбирают посторонние включения. Средние пробы образцов весом 30 г, подлежащие анализу, растирают в фарфоровой ступке и просеивают через сито 0,50 мм. Пробу квартуют и отбирают для анализа две параллельные пробы по 3-5 г. Если в пробе заведомо много нефтепродуктов, берут навеску 0,5-1,0 г (табл. IV.6.1).
IV.6.5. Проведение определения
IV.6.5.1. Предварительные указания. На точность определения нефтепродуктов в большой степени влияет чистота посуды и применяемых реактивов. Вся используемая при анализе посуда (воронки, колонки, колбы, мерные стаканы, бюксы) должна быть тщательно вымыта и освобождена от следов жира путем встряхивания с небольшим количеством четыреххлористого углерода. Реактивы и растворитель должны быть очищены и перегнаны.
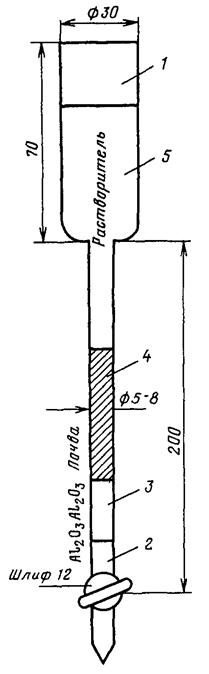
Рис.IV.6.1. Хроматографическая колонка:
1 - колонка, 2 - ватный тампон, 3 - алюминий окись (Аl2О3), 4 - почва, 5 - растворитель
Рис. IV.6.2. Штатив для колонок:
1, 2 - круги для размещения колонок и приемников для экстрактов соответственно; 3 - соединительные шпильки; 4 - крепежные болты; 5 - основание
Эффективность отделения нефтепродуктов зависит от способа подготовки окиси алюминия, поэтому следует обратить внимание на раздел IV.6.2.
IV.6.5.2. Мешающие влияния. На результаты определения могут оказывать влияние углеводороды неспецифических составляющих почвенного гумуса. К числу таких соединений относятся разнообразные физиологически активные вещества, углеводы, аминокислоты, различные пигменты, за которыми в настоящее время закрепился термин "липиды". Критерием оценки их вклада является фоновый уровень содержания неполярных и малополярных соединений в пробе почвы, взятой в месте этого же региона, не подергавшемся загрязнению. Литературные данные [6] свидетельствуют о том, что доля неполярных и малополярных углеводородов естественного происхождения в почвах разного типа колеблется от 0,04 до 1,0 г/кг, причем наиболее частыми значениями являются 0,20-0,05 г/кг.
IV.6.5.3. Экстракция нефтепродуктов и очистка экстрактов. Экстракцию нефтепродуктов из почвенного образца и очистку элюата от примесей полярных углеводородов осуществляют одновременно в хроматографической колонке [7]. Для этого готовят стеклянную колонку (1) (см. рис. IV.6.1), помещают в нее ватный тампон (2), насыпают навеску сорбента (Аl2О3) (3), смачивают его растворителем ССl4, засыпают навеску пробы (4), сверху заливают тем же растворителем (5). Процесс извлечения осуществляют при комнатной температуре при истечении элюата в приемник (мерный цилиндр или стакан) со скоростью 0,1-0,2 мл/мин. По мере прокалывания растворителя его доливают в колонку. Объем элюата, достаточный для количественного извлечения нефтепродуктов из почв с различным уровнем концентрации, указан в табл. IV.6.1, в которой приводятся оптимальные условия экстракции. Объем элюата при ИКС окончании тщательно замеряют.
Вклад в результаты анализа неконтролируемых факторов осуществляют с помощью постановки "пустого опыта". Для этого в каждой серии анализов ставят колонку, не содержащую пробу почвы, и пропускают 30 мл растворителя, как описано выше.
С целью интенсификации процесса экстракции стеклянные колонки рекомендуется располагать на штативе (рис. IV.6.2).
Штатив для колонок состоит из двух кругов, жестко скрепленных шпильками. В верхнем вырезаются специальные гнезда для закрепления колонок или просверливаются отверстия, если используются колонки без кранов на конце. Нижний круг предназначен для расположения приемников (мерных цилиндров). Вся система насаживается на ось для свободного вращения. Такого типа штатив может быть рассчитан на 15-20 колонок, что позволяет одновременно анализировать такое же количество проб.
Таблица IV.6.1
Оптимальные условия экстракции нефтепродуктов из почв в колонке при комнатной температуре
|
Уровень концентрации нефтепродуктов в почве, г/кг |
|||
|
1,0 |
1,0-20,0 |
20,0 |
|
|
Навеска, г |
3-5 |
3-5 |
0,5-1,0 |
|
Объем элюата, мл |
20-30 |
40-60 |
60-70 |
|
Количество сорбента для хроматографической очистки, г |
1-3 |
1-3 |
3-5 |
IV.6.6. ИК спектрометрический анализ экстрактов
IV.6.6.1. Приготовление стандартных растворов и построение калибровочных кривых. Для построения калибровочной кривой в качестве стандартного раствора используют смесь, состоящую из 2,5 мл изооктана, 5,0 мл н-гексадекана и 2,5 мл бензола; систематическая ошибка при этом не превышает 1,0 %. Полученную смесь растворяют в ССl4, в мерной колбе на 100 мл. Концентрация полученного раствора 77,8±0,8 мг/мл (раствор А). Из раствора А готовят два рабочих раствора Б. По 1,0 мл раствора (А) разбавляют в мерных колбах на 25 мл (раствор Б1) и на 100 мл (раствор Б2). Растворы Б1 и Б2 содержат 3,10±0,03 и 0,80±0,01 мг/мл соответственно и служат основными стандартными растворами. Две серии стандартных растворов с концентрациями 3,10; 0,60; 0,40; 0,20 и 0,10 мг/мл готовят последовательным разбавлением соответствующих основных растворов Б1 и Б2 четыреххлористым углеродом.
Одну кювету заполняют ССl4, предварительно пропущенным через колонку с окисью алюминия (кювета сравнения), другую - исследуемым раствором.
Измеряют интенсивность поглощения каждого раствора на инфракрасном спектрометре в интервале длин волн 2700-3100 см-1.
Измерения проводят в неразборных или разборных кюветах (для концентрации нефтепродуктов ниже 1,0 г/кг). Разборные кюветы состоят из двух окошек из кристаллов NaCl (прилагаются к спектрометру) и стеклянного цилиндра длиной 10 мм (прилагается к спектрофотометру СФ-4А(СФ-16) или можно вырезать из тефлона).
Оптическую плотность рассчитывают методом базисной линии. Последнюю проводят как касательную к основанию двух пиков, соответствующих симметричным и асимметричным валентным колебаниям СН2 и СН3 групп, как показано на рис. IV.6.3. Анализ ведут по полосе поглощения асимметричных валентных колебаний метиленовых групп (2926 см-1) либо по сумме оптических плотностей двух или трех длин волн [8].
На регистрирующей ленте указывают дату анализа, маркировку пробы, толщину кюветы и объем элюата.
Оптическую плотность вычисляют по формуле
E=lg I0/I, (IV.6.1)
где I0, I - интенсивность падающего и прошедшего через раствор излучения. При анализе нефтепродуктов по одной длине волны I измеряют в максимуме поглощения полосы aS - СН2 n=2926 см-1;
I0 - интенсивность потока в отсутствие поглощения.
Далее строят калибровочные графики, откладывая по оси ординат оптическую плотность, по оси абсцисс - концентрацию нефтепродуктов (мг/мл) в пробе. Вид калибровочного графика изображен на рис. IV.6.4.
Все стандартные растворы необходимо хранить только в стеклянной посуде с притертыми пробками при комнатной температуре в вытяжном шкафу.
IV.6.6.2. Обработка результатов анализа. Содержание (г/кг) нефтепродуктов в пробе (Х) при ИКС аналитическом окончании определяют по формуле (IV.5.1)
![]() (IV.6.2)
(IV.6.2)
где С - концентрация нефтепродуктов в пробе, найденная по калибровочному графику, мг/мл; V - объем исходного элюата, мл; m - навеска пробы, г.
IV.6.6.3. Метрологические характеристики. Вариационно-статистические характеристики ИК спектрометрического метода анализа почв на содержание в них нефтепродуктов представлены в табл. IV.6.2.
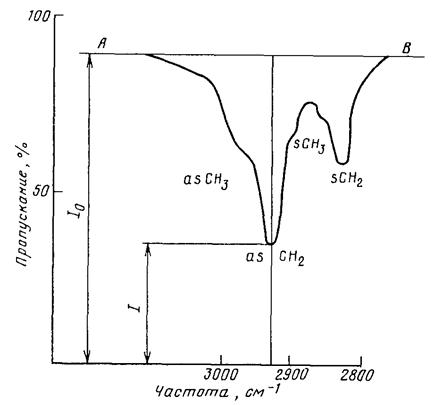
Рис. IV.6.3. Измерение интенсивности поглощения раствора нефтепродуктов методом базовой линии:
АВ - базовая линия; I0, I - интенсивность падающего и прошедшего через раствор излучения
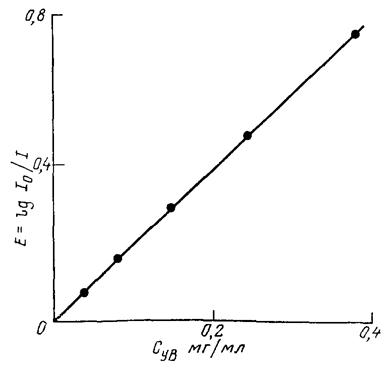
Рис. IV.6.4. Калибровочный график для определения концентрации нефтепродуктов
Таблица IV.6.2
Вариационно-статистические характеристики анализа нефтепродуктов в почве ИКС методом
|
Интервал концентрации нефтепродуктов в почве, г/кг |
|||
|
0,05-1,00 |
1,00-20,0 |
>20,0 |
|
|
Среднее арифметическое |
0,09 |
2,77 |
69,5 |
|
Среднее квадратичное отклонение SХ |
4,60×10-2 |
0,28 |
4,4 |
|
Коэффициент вариации V % |
51,10 |
10,10 |
6,3 |
|
Стандартное среднее квадратичное отклонение D |
0,96×10-2 |
5,61×10-2 |
0,88 |
|
Относительная ошибка Q % при доверительной вероятности Р = 0,95 |
22,0 |
4,1 |
2,6 |
|
Объем выборки n |
22 |
30 |
25 |
Список литературы
2. Добыча полезных ископаемых и геохимия экосистем. -М.: Наука. 1982. - 278 с.
3. Люминесцентная битуминология/ Под ред. В.Н. Флоровской. - М.: Изд-во МГУ, 1975. - 192 с.
4. Техногенные потоки вещества в ландшафтах и состояние экосистем. - М.: Наука, 1981. - 256 с.
5. Охрана окружающей среды в нефтедобывающей промышленности. М.: Химия, 1980. - 174 с.
9. Полевое обследование и картографирование уровня загрязнения почвенного покрова техногенными выбросами через атмосферу (Методические указания). - М.: ВАСХНИЛ, 1980. - 26 с.
РАЗДЕЛ V. МЕТОДИЧЕСКИЕ УКАЗАНИЯ ПО ИСПОЛЬЗОВАНИЮ ФЕРМЕНТАТИВНЫХ ПОКАЗАТЕЛЕЙ ДЛЯ ОЦЕНКИ ПОСЛЕДСТВИЙ ЗАГРЯЗНЕНИЯ ПОЧВ
V.1. Введение
Все возрастающее загрязнение окружающей среды создает угрозу стойкого и необратимого изменения химического состава, физических, биохимических и микробиологических свойств почвы, определяющих ее плодородие. Для оценки состояния почвы в измененных и изменяющиеся условиях окружающей среды основное значение приобретают не количественные характеристики загрязнений сами по себе, а их последствия.
Почва подвергается интенсивному антропогенному влиянию и служит одним из опасных звеньев циркуляции промышленных и сельскохозяйственных токсических веществ. Разнообразные химические реакции в почве, связанные с обменом веществ, разложением и синтезом органических веществ, миграцией химических соединений, мобилизацией питательных элементов и т.д., осуществляются ферментативно. Высокая чувствительность, точность, относительная простота и нетрудоемкость методов определения активности почвенных ферментов позволяют использовать их при оценке интенсивности и направленности важнейших для жизни и плодородия почвы биохимических процессов. По активности ферментов судят об агрономически значимых показателях, плодородии, превращении гумусовых веществ, окислительно-восстановительном режиме почвы. Активность ферментов отражает интенсивность основных биохимических процессов: самоочищения почвы и разложения органических соединений азота, фосфора, углерода, а также степень эродированности и загрязнения почв [1-3].
Биодиагностика состояния и степени загрязнения почв по ферментативным показателям приобрела в последнее время особо важное значение [9]. Отсутствие стандартных, унифицированных методов определения активности почвенных ферментов затрудняет выявление возможности и области использования ферментов в целях биодиагностики.
V.2. Выбор показателей для оценки загрязнения почв
Использование ряда ферментативных показателей при оценке общей биологической активности и плодородия почвы является в настоящее время общепринятым [4, 5]. В качестве диагностического показателя загрязнения почв наиболее перспективными оказались ферменты класса оксидоредуктаз, в частности дегидрогеназы. Показана высокая корреляция активности дегидрогеназ в почве с активностью протеаз, интенсивностью процессов нитрификации, азотфиксации, дыхания (по СО2), поглощением почвой кислорода и плодородием. Высокая чувствительность дегидрогеназ к химическим веществам используется при оценке токсичности сточных вод промышленных предприятий. Все сказанное послужило основанием при выборе активности дегидрогеназ в качестве одного из диагностических показателей загрязнения почв.
Учитывая то, что окислительно-восстановительные и гидролитические процессы в почве протекают сопряженно и часть энергии, образованной в одних реакциях, используется в других, при биодиагностике загрязнения почв необходимо проводить определение активности гидролитических ферментов. Особого внимания среди гидролаз заслуживают фосфатаза, с помощью которой осуществляется метаболизм фосфора в почве, и уреаза, с действием которой связано превращение азотсодержащих соединений в почве. Высокая чувствительность уреазы и фосфатазы к действию металлов и пестицидов послужила основанием для выбора этих показателей в качестве индикаторов загрязнения почв. Продуцирование СО2 является результатом сопряженно протекающих гидролитических и окислительно-восстановительных процессов, которые в основном определяются биологическими факторами. "Дыхание" почвы (выделение CO2) выбрано как интегральный показатель, характеризующий действие оксидаз (дегидрогеназы) и гидролаз (фосфатазы и уреазы).
V.3. Отбор и подготовка почвенных образцов к анализу
Методика отбора почвенных образцов определяется поставленными перед исследователем задачами. При исследовании влияния антропогенных загрязнений на активность почвенных ферментов в промышленных и сельскохозяйственных районах отбор почвенных образцов проводится на глубине пахотного слоя (0-5-7 см). Ферментативная активность почв значительно уменьшается с глубиной, что необходимо иметь в виду при отборе проб. При определении ферментативной активности на каждую стометровую делянку рекомендуют брать пять образцов [6]. Каждый образец составляют из трех смешанных проб. Если делянка меньше 100 м, то достаточно брать три образца по диагонали, составленные из трех смешанных проб. Каждый образец анализируют отдельно.
Для анализов почву тщательно очищают от корней и других растительных остатков и прочих включений, высушенные пробы растирают и просеивают через сито с диаметром отверстий 2 мм.
Во всех случаях показатели ферментативной активности переводят на вес воздушно-сухой или абсолютно сухой почвы и обязательно указывают, какие образцы почвы (сухие или естественно-влажные) проанализированы.
V.4. Определение активности дегидрогеназ [5, 7]
1 г воздушно-сухой почвы, а влажной с учетом влажности, помещают в стеклянную пробирку и поливают 2 мл 0,5%-ного водного раствора ТТХ, суспензию тщательно перемешивают и помещают в термостат на 24 ч при температуре 30 °С. После инкубации смесь центрифугируют в течение 5 мин при 3000 об/мин, надосадочную жидкость сливают. Для извлечения формазана, образовавшегося в процессе восстановления ТТХ, почву заливают 7,5 мл ацетона. Экстракцию проводят в течение 1 ч. Затем пробирки со смесью тщательно встряхивают и центрифугируют 5 мин при 3000 об/мин. Окрашенный раствор сливают в пробирки и колориметрируют на ФЭК с синим светофильтром, используя кюветы толщиной 5 или 1 мм в зависимости от концентрации растворов.
Количество формазана рассчитывают по калибровочной кривой (рис.V.4.1) и выражают в микролитрах водорода (мкл Н2/г почвы.ч) с учетом того, что на образование 1 мг формазана необходимо 150, 35 мкл водорода.
Результаты измерений активности дегидрогеназ в чистых и загрязненных образцах характеризуют соответствующие состояния почвенной микрофлоры.
Расчет активности дегидрогеназ ведется по формуле
![]() (V.4.1)
(V.4.1)
где С - концентрация формазана, найденная по калибровочной кривой; а - навеска почвы (1 г); Т - время инкубации (24 ч); 10 мл - объем формазана, используемый при построении калибровочной кривой; в - количество ацетона, пошедшее на извлечение формазана (7,5 мл).
На основании закона Бера С можно выразить как
![]() (V.4.2)
(V.4.2)
где С - концентрация поглощающегося вещества; D - показания ФЭК, характеризующие поглощение света; К1 - коэффициент пропорциональности.
Подставляя это выражение в формулу для определения активности дегидрогеназ, получают
![]() (V.4.3)
(V.4.3)
При использовании кюветы толщиной 1 мм
![]() (V.4.4)
(V.4.4)
Результаты фотоэлектроколориметрирования и вычисления АД записывают по образцу, представленному в табл. V.4.1.
Таблица V.4.1
Результаты определения активности дегидрогеназ
|
Вариант |
Кювета (l) |
Показания ФЭК (D) |
АД |
Средняя АД |
Примечание |
|
|
1 марта 1983 г. |
2 |
1 |
0,049 |
0,90 |
0,79 |
|
|
0,039 |
0,72 |
|||||
|
0,040 |
0,74 |
|||||
|
0,044 |
0,81 |
V.4.1. Приготовление стандартных растворов и построение калибровочных кривых
Стандартный раствор ТФФ. 25 мг ТФФ взвешивают на аналитических весах, растворяют в 100 мл ацетона при нагревании до 30 °С на водяной бане. Концентрация полученного основного раствора 0,25 мг/мл. Раствор готовят непосредственно перед употреблением.
Стандартные рабочие растворы ТФФ готовят из основного раствора, разводя его в 2, 5, 10, 20, 50 и 100 раз ацетоном. Для этого в градуированные пробирки помещают 5; 2; 1; 0,5; 0,2; 0,1 мл основного раствора ТТФ и доливают до 10 мл ацетоном. Содержимое пробирок тщательно перемешивают и оставляют на 5 мин. Каждая концентрация готовится в трех-четырех повторностях. В течение 1 ч после приготовления рабочие растворы должны быть проанализированы, при этом не следует допускать попадания на них прямых солнечных лучей. Интенсивность окраски рабочих растворов ТФФ, содержащих от 0,0025 до 0,1250 мг/мл вещества, измеряют на ФЭК с синим светофильтром, используя кювету толщиной 1 мм.
Результаты измерения оптической плотности рабочих растворов приведены в табл. V.4.2. Калибровочная кривая, построенная по этим данным, представлена на рис. V.4.1. Линейная зависимость оптической плотности от концентрации сохраняется в интервале от 0,0025 до 0,1250 мг/мл.
Таблица V.4.2
Результаты измерения оптической плотности (Х±S) рабочих растворов ТФФ
|
Концентрация ТФФ, мг/мл |
Средняя оптическая плотность D |
|
|
1 |
0,0025 |
0,0088±0,0015 |
|
0,0050 |
0,0163±0,0026 |
|
|
0,0125 |
0,0330±0,0053 |
|
|
0,0250 |
0,0620±0,0014 |
|
|
0,0500 |
0,1213±0,0092 |
|
|
0,1250 |
0,3045±0,0098 |
Значение средней оптической плотности определяли по формуле
![]()
а значение S (оценка среднего квадратичного отклонения) - по формуле
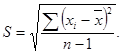
Оценки получены при n=4.
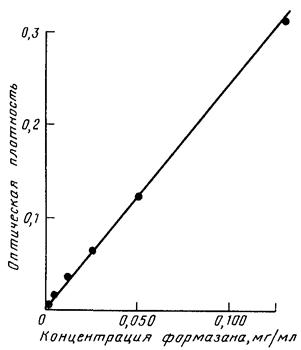
Рис. V.4.1. Калибровочный график для определения концентрации формазана
V.4.2. Приготовление рабочих растворов
0,5 %-ный раствор 2,3,5-ТТХ. 0,5 г 2,3,5-ТТХ растворяют в 100 мл дистиллированной воды. Раствор готовят перед употреблением.
Основной раствор ТФФ. 25 мг ТТФ растворяют в 100 мл ацетона при нагревании до 30°С на водяной бане.
V.4.3. Реактивы
Ацетон ч.д.а., ГОСТ 2606-63.
Трифенил - третразолий хлористый (2,3,5 - ТТХ) ч.д.а. ТУ 6-09-5328-68.
Трифенилформазан (формазан: 2,3,5 - ТФФ) получают следующим образом. 2,3,5 - ТТХ растворяют в 5 мл дистиллированной воды. 1,26 г гидросульфита натрия (NaHSO3) растворяют в 5 мл дистиллированной воды. Растворы сливают, после выпадения темно-красного осадка смесь фильтруют через стеклянный пористый фильтр и осадок промывают на фильтре дистиллированной водой до отсутствия реакции на хлориды (с 0,5%-ным раствором AgNО3). Осадок (2,3,5 - трифенилформазан) высушивают в сушильном шкафу.
Гидросульфит натрия ч.д.а. ТУ МХПГХП 126-56.
Азотнокислое серебро ч.д.а. ГОСТ 1277-63.
V.5. Определение активности фосфатазы (АФ) [5]
Навеску почвы (1 г воздушно-сухой почвы, а влажной - с учетом влажности) помещают в пробирку и заливают 2 мл 1%-ного раствора фенолфталеинфосфата натрия, рН которого доводят до 7,0. Пробирки закрывают резиновыми пробками, тщательно взбалтывают и помещают в термостат на 3 ч при 30 °С. После инкубации для извлечения продукта гидролиза органического субстрата (фенолфталеина) и получения бесцветных и прозрачных почвенных вытяжек в каждую пробирку добавляют 5 мл 0,1 М СаСl2 или 0,1 % К2SO4, хорошо перемешивают и центрифугируют в течение 10 мин при 3000 об/мин. Надосадочную жидкость сливают в чистые пробирки.
К 5 мл центрифугата приливают 5 мл 10%-ного раствора аммиака. Окрашенный в розовый цвет раствор колориметрируют на ФЭК с синим светофильтром, используя кювету толщиной 20 мм. Окраска устойчива в течение 1 ч. Активность фосфатазы выражают в мг фенолфталеина на 1 г воздушно-сухой почвы за 1 ч (мг/г ч). Количество фенолфталеина, отщепленного от фенолфталеинфосфата натрия под действием фосфатазы, находят по калибровочной кривой.
Расчет активности фосфатазы ведется по формуле
![]() (V.5.1)
(V.5.1)
где С - концентрация фенолфталеина, найденная по калибровочной кривой; V1 - количество раствора для приготовления почвенной вытяжки (7 мл); V2 - аликвота (5 мл); а - навеска почвы (1 г); Т - время инкубации (3 ч).
На основании закона Бера С можно выразить как
![]() (V.5.2)
(V.5.2)
где С - концентрация поглощающего вещества; D - показания ФЭК, характеризующие поглощение света; К1 - коэффициент пропорциональности.
Подставляя это выражение в формулу для определения активности фосфатазы, получают
![]() (V.5.3)
(V.5.3)
При использовании кюветы толщиной 20 мм
![]() (V.5.3а)
(V.5.3а)
Результаты фотоэлектроколориметрирования и вычисления АФ записывают по образцу, представленному в табл. V.5.1.
Таблица V.5.1
Пример записи результатов вычисления АФ
|
Вариант |
Толщина кюветы, мм |
Показания ФЭК (D) |
АФ |
Средняя АФ |
Примечание |
|
|
1 марта 1983 г. |
1 |
20 |
0,115 |
0,10 |
||
|
0,109 |
0,10 |
|||||
|
0,120 |
0,11 |
0,10 |
||||
|
0,118 |
0,10 |
|||||
|
2 |
0,200 |
0,18 |
0,18 |
|||
|
0,205 |
0,18 |
|||||
|
0,200 |
0,18 |
|||||
|
0,195 |
0,17 |
V.5.1. Приготовление стандартных растворов фенолфталеина и построение калибровочной кривой
0,5 г фенолфталеина растворяют в 100 мл этанола, получают раствор 0,5 %-ной концентрации. Из стандартного раствора берут аликвоты, содержащие 0,1; 0,2; 0,5; 1,0; 2,0; 5,0 мг фенолфталеина, и переносят в мерные колбы емкостью 50 мл, доливают этанолом до метки. Получают таким образом серию рабочих растворов, 1 мл которых содержит 0,002; 0,004; 0,01; 0,02; 0,04; 0,1 мг фенолфталеина, 5 мл рабочего раствора переносят в пробирку и добавляют 5 мл 10 %-ного раствора аммиака. Окрашенные растворы колориметрируют на ФЭК с синим светофильтром. Окраска устойчива в течение 1 ч.
Результаты измерения оптической плотности окрашенных растворов фенолфталеина различных концентраций представлены в табл. V.5.2, калибровочная кривая для кюветы толщиной 20 мм - на рис. V.5.1. Линейная зависимость оптической плотности от концентрации растворов фенолфталеина наблюдается в интервале от 0,01 до 0,5 мг/10 мл.
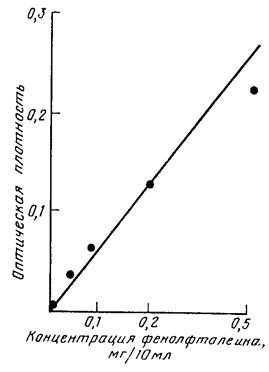
Рис. V.5.1. Калибровочная кривая для определения концентрации фенолфталеина
Таблица V.5.2
Результаты измерения оптической плотности рабочих растворов фенолфталеина
|
Концентрация фенолфталеина, мг ФФ в 10 мл |
Средняя оптическая плотность |
|
|
20 |
0,01 |
0,012 |
|
0,02 |
0,022 |
|
|
0,05 |
0,037 |
|
|
0,1 |
0,065 |
|
|
0,2 |
0,110 |
|
|
0,5 |
0,227 |
V.5.2. Приготовление рабочих растворов
0,1%-ный раствор фенолфталеина. 100 мг фенолфталеина растворяют в 100 мл 60%-ного этилового спирта. Раствор перемешивают.
1%-ный раствор фенолфталеинфосфата натрия. 10 мл 10%-ного раствора (торговый препарат) разбавляют дистиллированной водой до 80 мл, затем концентрированной соляной кислотой, разбавленной водой в отношении 1:2,5, доводят pH раствора до 7,0. Объем раствора дистиллированной водой доводят до 100 мл. Раствор готовят перед употреблением.
0,1 М СаСl2. 11 г обезвоженной соли СаСl2 помещают в мерную колбу на 1 л, растворяют в небольшом объеме дистиллированной воды, доводят водой до метки и перемешивают.
10%-ный раствор NH4OH. 30%-ный раствор аммиака разбавляют дистиллированной водой в три раза. Раствор хранят в холодильнике.
V.5.3. Реактивы
Фенолфталеин индикатор ч.д.а.
Фенолфталеинфосфат натрия (ФФФ Nа) 10%-ный раствор ч., ТУ 6-09-2961.
Хлористый кальций (СаСl2) х.ч., ГОСТ 4460-66.
Соляная кислота (HCl) пл. - 1,12, ГОСТ 3118-67, х.4.
Аммиак (NH4OH) 30% пл.- 0,91 ч., ГОСТ 3760-64.
Этиловый спирт 60 %.
V.6. Определение активности уреазы (АУ) [5]
Навеску почвы (1 г воздушно-сухой почвы, а влажной - с учетом влажности) помещают в пробирку и заливают 2 мл 2%-ного раствора мочевины. Для контроля берут пробирку с такой же навеской почвы и добавляют к ней 2 мл дистиллированной воды. Пробирки закрывают резиновыми пробками, тщательно встряхивают и ставят в термостат на 3 ч при 30 °С. После инкубации в каждую пробирку доливают по 8 мл дистиллированной воды и для получения прозрачных растворов по две капли разведенной соляной кислоты (1:2).
Суспензию взбалтывают и центрифугируют при 3000 об/мин в течение 3 мин. Для определения количества аммиачного азота в реакционной смеси 1 мл центрифугата помещают в мерную колбу на 50 мл, доливают водой примерно до 10 мл, затем прибавляют 4 мл раствора фенолята натрия и сразу же 3 мл раствора гипохлорита натрия. После добавления каждого реактива смесь тщательно перемешивают. Через 20 мин объем смеси в колбе доводят до метки. Окрашенные в голубой цвет растворы колориметрируют на ФЭК с красным светофильтром. Окраска индофенола голубого устойчива в течение 1 ч.
Активность уреазы выражают в мг N-NH3/г.ч. Количество аммиачного азота находят по стандартной калибровочной кривой (рис. V.6.1). Расчет количеств аммиачного азота ведется по формуле
![]() (V.6.1)
(V.6.1)
где x - количество аммиачного азота (N-NH3, мг); С - концентрация N-NH3, найденная по калибровочной кривой; V1 - количество раствора для приготовления почвенной вытяжки (10 мл); V2 - аликвота (1 мл); а - навеска почвы (г); Т - время инкубации (3 ч); 1000 - коэффициент для перевода мкг в мг.
На основании закона Бера С можно выразить
![]()
где С - концентрация поглощающего вещества; D - показание ФЭК, характеризующее поглощение света; К1 - коэффициент пропорциональности.
Подставляя это выражение в формулу для вычисления количества аммиачного азота N-NH3(x) , получают
![]() (V.6.2)
(V.6.2)
При использовании кюветы толщиной 20 мм имеют
![]() (V.6.3)
(V.6.3)
Активность уреазы равна разнице между опытными и контрольными значениями
АУ=Х0-ХК.
Рис. V.6.1. Калибровочная кривая для определения концентрации N-NН3
Результаты фотоэлектроколориметрирования и вычисления АУ записывают по образцу, представленному в табл. V.6.1.
Таблица V.6.1
Пример записи результатов определения активности уреазы
|
Вариант |
Кювета |
Показания ФЭК (D) |
Количество N-NH3, мг |
АУ |
Средняя АУ |
Примечание |
|
|
1 марта 1983 г. |
1 |
20 |
0,209 |
0,05 |
0,03 |
0,03 |
|
|
0,208 |
0,05 |
0,03 |
|||||
|
0,205 |
0,05 |
0,03 |
|||||
|
0,211 |
0,05 |
0,03 |
|||||
|
0,080 |
0,02 |
||||||
|
0,086 |
0,02 |
V.6.1. Приготовление стандартного раствора сернокислого аммония и построение калибровочной кривой
0,4717 (NH4)2SO4, высушенного до постоянного веса, помещают в мерную колбу емкостью 1000 мл, растворяют в дистиллированной воде и объем раствора доводят до метки.
Концентрация стандартного раствора равна 100 мкг N/мл. Для приготовления рабочих растворов в мерные колбочки емкостью 100 мл отбирают 1, 2, 5, 8, 12, 15, 18, 22, 25, 28 мл исходного раствора и доливают дистиллированной водой до метки. В 1 мл каждого раствора содержится соответственно 1, 2, 5, 8, 12, 15, 18, 22 мкг N-NH3.
1 мл рабочего раствора помещают в мерную колбу емкостью 50 мл, доводят объем дистиллированной водой до 10 мл, затем приливают 4 мл раствора фенолята натрия и сразу же 3 мл раствора гипрохлорита натрия. После добавления каждого реактива смесь тщательно перемешивают. Через 20 мин объем смеси доводят до метки и интенсивность окраски просматривают, на ФЭК с красным светофильтром, используя кювету толщиной 20 мм. Окраска индофенола голубого устойчива в течение 1 ч. Результаты измерений оптической плотности представлены в табл. V.6.2, калибровочная кривая - на рис. V.5.1. Линейная зависимость оптической плотности от концентрации N-NH3 для кюветы толщиной 20 мм сохраняется в интервале от 1 до 22 мкг/50 мл.
Таблица V.6.2
Результаты измерений оптической плотности рабочих растворов (NH4)2SO4
|
Концентрация аммиачного азота, мкг N-NH3 в 50 мл |
Средняя оптическая плотность |
|
|
20 |
1 |
0,019 |
|
2 |
0,036 |
|
|
5 |
0,061 |
|
|
8 |
0,104 |
|
|
12 |
0,154 |
|
|
15 |
0,199 |
|
|
18 |
0,221 |
|
|
22 |
0,270 |
V.6.2. Приготовление рабочих растворов
2%-ный раствор мочевины. 2 г мочевины растворяют в 100 мл дистиллированной воды, перемешивают. Раствор готовят перед употреблением.
Раствор фенолята натрия. 62,5 г фенола растворяют в малом количестве этанола, добавляют 18,5 мл ацетона, этанолом доводят объем раствора до 100 мл, перемешивают (раствор А). 27 г NaOH растворяют в 100 мл дистиллированной воды (раствор Б). Оба раствора хранят в холодильнике. Перед употреблением по 20 мл растворов А и Б смешивают, объем доводят до 100 мл и перемешивают.
Раствор гипохлорита натрия. Исходный препарат разбавляют водой до концентрации активного хлора, равной 0,9 %. К приготовленному исходному раствору гипохлорита натрия прибавляют дистиллированную воду в отношении 1:10. Раствор устойчивый.
Разбавленная соляная кислота (1:2). К 20 мл дистиллированной воды осторожно приливают 10 мл концентрированной соляной кислоты, перемешивают.
V.6.3. Реактивы
Мочевина (CH4ON2) ч.д.а., ГОСТ 6691-67.
Фенол (С6Н5ОН ) ч., ГОСТ 6417-52.
Спирт этиловый ректификат (96,4%), ГОСТ 18300-72.
Едкий натр (NaOH) ч.д.а., ГОСТ 4328-66.
Соляная кислота (HCl) пл.- 1,12 х.ч., ГОСТ 3118-67.
Углекислый натрий безводный (Na2CO3) ч.д.а., ГОСТ 83-63.
Хлорная известь (имеющаяся в продажа).
Раствор гипохлорита натрия (натрий хлоноватистокислый, NaClO) получают следующим образом. Тщательно размешивают 100 г хлорной извести c содержанием активного хлора 35-36 % с 170 мл дистиллированной воды в течение 15 мин и в смесь при непрерывном перемешивании вносят раствор Na2CO3 (70 г в 170 мл дистиллированной воды). Масса сначала густеет, затем опять разжижается. Жидкость отделяют от осадка фильтрованием через полотняный фильтр на воронке Бюхнера. Получается 320 мл раствора NaClO с содержанием активного хлора 71-100 г/л.
V.7. Определение интенсивности "дыхания" почвы [8]
Методика основана на измерении количества СО2, выделившегося из почвы за определенный промежуток времени, и состоит в следующем. Почву площадью 60-80 см2 изолируют с помощью стеклянного или полиэтиленового сосуда, края которого заглубляют на 1,5-2 см. Внутри этого сосуда на пластмассовой подставке устанавливают стеклянную чашечку с 5 мл поглощающей щелочи и выдерживают 2 ч. В качестве поглотителя СО2, выделившегося из почвы, используют раствор 0,1 н КОН.
Для определения количества углекислоты, содержащейся в воздухе сосуда-изолятора, такую же чашечку с 5 мл щелочи устанавливают на подставке в чашке Петри, заполненной слегка подкисленной водой, закрывают сосудом и выдерживают 2 ч.
Интенсивность выделения СО2 почвой определяется по формуле
![]() (V.7.1)
(V.7.1)
где D - количество СО2, выделившегося из почвы, мг/дм2/ч;
а - количество 0,1 н HCl, которое пошло на титрование щелочи при определении содержания СО2 в воздухе сосуда-изолятора, мл:
b - количество 0,1 н HCl , которое пошло на титрование щелочи в опыте, мл;
S - площадь изолируемой поверхности, дм2;
Т - время экспозиции;
2,2 - количество СО2, поглощаемое 1 мл 0,1 н раствора щелочи, мг.
Например, при 2-часовой экспозиции на титрование 5 мл щелочи при определении содержания СО2 в воздухе сосуда-изолятора пошло 4,92 мл 0,1 н HCl, а на титрование щелочи в опытном варианте - 4,92 мл 0,1 н НСl. Тогда интенсивность выделения СО2 из почвы площадью 0,665 дм2 за 2 ч, поглощенного 5 мл щелочи, будет равна
![]() мг
СО2/дм2ч (V.7.2)
мг
СО2/дм2ч (V.7.2)
Чем больше поглотилось углекислоты щелочью, тем меньше количество 0,1 н HCl пойдет на ее титрование, так как часть щелочи уже будет нейтрализована углекислотой.
V.7.1. Приготовление рабочих растворов
0,1 н раствор КОН готовят из фиксанала, тщательно перенося его содержимое в мерную колбу емкостью 1 л, ампулу, боек и воронку при этом тщательно ополаскивают дистиллированной водой. Объем раствора в колбе доводят до метки. Приготовленный раствор щелочи хранят в холодильнике с хлоркальциевой трубкой.
0,1 н раствор HCl готовится из фиксанала.
1%-ный раствор фенолфталеина. 1 г фенолфталеина растворяют в 100 мл этилового спирта (60 %).
V.7.2. Реактивы
Фиксанал щелочи (0,1 н КОН).
Фиксанал кислоты (0,1 н НСl).
Фенолфталеин индикатор ч.д.а.
Спирт этиловый 60 %.
Список литературы
2. Симонян Б.Н., Галстян А.Ш. Диагностика эродированных почв по активности ферментов. - В кн.: Проблемы и методы биологической диагностики и индикации почв. М.: Наука, 1976.- 400 с.
3. Долгова Л.Г. Применение ферментативной активности как одного из диагностических показателей, характеризующих загрязнение промышленными выбросами почвы. - В кн.: Биологическая диагностика почв. М.; Наука, 1976, с.76-77.
4. Галстян А.Ш. Ферментативная активность почв Армении. Ереван: Айстен, 1974.- 257 с.
5. Хазиев Ф.Х. Ферментативная активность почв. - М.: Наука, 1976.- 179 с.
8. Методы стационарного изучения почв. - М.: Наука, 1977.
9. Краснова Н.М. Ферментативная активность как биоиндикатор загрязнения почв тяжелыми металлами. Автореф. дисс. на соиск. учен. степени канд. биол. наук. -М., ВАСХНИЛ, 1982.- 25 с.
РАЗДЕЛ VI. МЕТОДЫ ОПРЕДЕЛЕНИЯ НЕКОТОРЫХ АГРОХИМИЧЕСКИХ ПОКАЗАТЕЛЕЙ ПОЧВЫ
В практике осуществления мониторинга загрязнения почв при оценке последствий загрязнения, особенно при оценке влияния токсичных загрязнений на наиболее важные физико-химические свойства почв, может возникнуть необходимость определения характеристики этих свойств или тенденций изменения их во времени. Ниже приводятся методики определения некоторых наиболее важных агрохимических свойств почвы, которые могут при этом быть полезными. К ним относятся методики определения в почве углерода, подвижного фосфора, нитратного азота, аммиачного азота, сульфатов. Как правило, для оценки последствий загрязнения указанные агрохимические характеристики на загрязненных участках почвы сравниваются с такими же характеристиками на аналогичных контрольных участках, не подверженных загрязнению (с фоном).
VI.1. Определение содержания общего углерода в почве [1-3]
Навеску почвы, подготовленную путем тщательного отбора растительных корешков и растирания в ступке до полного просеивания через сито с отверстиями диаметром 0,25 мм, отвешивают на аналитических весах в количестве 0,05-0,5 г в зависимости от содержания гумуса.
Навеску помещают в сухую узкогорлую коническую колбочку емкостью 100 мл. Определение проводят в двух-четырех повторностях. С помощью мерного цилиндра в колбочку с навеской почвы приливают 20 мл окислительной смеси. Содержимое колбы осторожно и тщательно перемешивают круговым движением. Колбочки закрывают маленькой воронкой для охлаждения паров и ставят на горячую электроплитку с закрытой спиралью или на песочную баню и кипятят точно 5 мин с начала момента кипения (появление первого относительно крупного пузырька газа).
Смесь охлаждают и доводят ее объем дистиллированной водой до 100 мл, затем тщательно перемешивают и оставляют на ночь для развития более устойчивой окраски. Отстоявшийся раствор осторожно (не взмучивая осадка) сливают в кювету спектрофотометра толщиной 1 см и измеряют оптическую плотность при длине волны 590 нм. Контролем служит прокипяченный и разбавленный (до 100 мл) раствор окислительной смеси.
Расчет содержания углерода ведется по формуле (при использовании кюветы толщиной 1 см):
![]()
где D - оптическая плотность, m - навеска почвы, г; С - содержание углерода, % к воздушно-сухой почве.
Для вычисления количества гумуса в почве нужно учитывать, что среднее содержание углерода в гумусе составляет 58 %, т.е.:
% гумуса = % С 1,72.
Присутствие хлора в почве искажает результаты анализа. Предельное его содержание, начинающее влиять на точность метода - 0,5-0,6 %.
VI.1.1. Определение содержания воднорастворимого органического вещества [1-3]
20 г воздушно-сухой почвы, а влажной - с учетом влажности, заливают 100 мл дистиллированной воды, соотношение 1:5, взбалтывают и оставляют на сутки. Затем снова взбалтывают, центрифугируют 15 мин при 3000 об/мин и фильтруют через плотный бумажный фильтр (синяя лента). Из фильтрата берут аликвоту 10-50 мл и выпаривают ее на электрической плитке с закрытой спиралью досуха. Затем проводят те же операции, что и при определении общего содержания органического вещества почвы.
Содержание углерода водорастворимого органического вещества в % к воздушно-сухой почве находят по формуле
![]()
где D - оптическая плотность; m - навеска воздушно-сухой почвы, соответствующая аликвоте, взятой для анализа (2-10 г); % С - процентное содержание растворимого органического углерода в почве.
Содержание углерода воднорастворимого органического вещества обычно выражают в процентах к СОБЩ.
VI.1.2. Приготовление рабочих растворов
Окислительная смесь: 0,4 н раствор К2Сr2О7, в разбавленной (1:1) серной кислоте. Берут 20 г тонко измельченного в фарфоровой ступке кристаллического К2Сr2О7, растворяют примерно в 200-300 мл дистиллированной воды (можно с подогреванием) и фильтруют через бумажный фильтр в мерную колбу емкостью 0,5 л. Объем раствора доводят до метки дистиллированной водой и переливают в термостойкий или фарфоровый стакан.
К этому раствору (под тягой) приливают небольшими порциями (примерно по 100 мл) 0,5 л H2SO4, (плотность 1,84) при осторожном и многократном перемешивании. Раствор закрывают стеклом и оставляют стоять до полного охлаждения. Затем перемешивают и переливают в бутыль или склянку с притертой пробкой. Хранят раствор в темноте.
VI.1.3. Реактивы
Серная кислота пл 1.84 х.ч., ГОСТ 4204-66.
Калия бихромат (К2Сr2О7) х.ч., ГОСТ 4220-65.
VI.2. Определение подвижного фосфора в почве [2-4]
2 г воздушно-сухой почвы, а влажной с учетом влажности, помещают в плоскодонные колбы емкостью 100 мл, заливают 20 мл 0,2 М раствора HCl, перемешивают 5 мин с помощью электромеханической мешалки, настаивают 24 ч. Затем надосадочную жидкость сливают в пробирки и центрифугируют 5 мин при 3000 об/мин.
Аликвоты почвенной вытяжки, содержащей от 0,003 до 0,04 мг Р2О5, переносят в мерные пробирки на 20 мл с притертыми стеклянными пробками.
При аликвоте, равной 1 мл, в пробирки доливают 7 мл дистиллированной воды, взбалтывают. Затем в пробирку приливают 2 мл кислого раствора молибденово-кислого аммония. Раствор перемешивают и прибавляют к нему 1 каплю восстановителя - 2,5 %-ного раствора SnCl2. Немедленно вновь перемешивают. Спустя 5-7 мин плотность окрашенных в синий цвет растворов определяют с помощью фотоэлектроколориметра, используя красный светофильтр (или длину волны 650 нм). Количество Р2О5 находят по стандартной калибровочной кривой, расчет ведут по формуле
![]() (VI.2.1)
(VI.2.1)
где Х - количество подвижного фосфора на 100 г воздушно-сухой почвы, мг Р2O5/100 г; С - концентрация Р2О5, найденная по калибровочной кривой, мг/кг; V1 - общий объем вытяжки, мл; V2 - объем аликвотной части, мл; а - навеска воздушно-сухой почвы, г; 100 - коэффициент пересчета на 100 г воздушно-сухой почвы; К - коэффициент пересчета на почву, высушенную при 100-105 °С.
VI.2.1. Приготовление стандартных растворов фосфата и построение калибровочной кривой
0,1917 г КН2РО4 х.ч. растворяют в дистиллированной воде в мерной колбе емкостью 1 л, доливают водой до метки, перемешивают, хранят в склянке из темного отекла. 1 мл полученного образцового раствора содержит 0,1 мг P2O5. Затем готовят серию рабочих растворов (в 3-4 повторностях), содержащих в 1 мл 0,001; 0,002; 0,004; 0,006; 0,008; 0,01 мг. Для этого из образцового раствора берут аликвоты соответственно 0,5; 1; 2; 3; 4; 5 мл, содержащие 0,5; 0,1; 0,2; 0,3; 0,4; 0,5 мг и переносят в мерные колбочки на 50 мл, объем которых затем доводят дистиллированной водой до метки. Затем их взбалтывают. Из каждой колбочки берут аликвоту 1 мл и переносят в пробирку (на 10-15 мл). Приливают 2 мл кислого раствора молибденово-кислого аммония (концентрация его в испытуемом растворе должна быть 0,001 мг/мл), затем 7 мл дистиллированной воды, перемешивают и капают 1 каплю восстановителя (2,5 % раствор SnCl2), немедленно перемешивают. Окрашенные в синий цвет растворы колориметрируют на ФЭКе (длина волны 650 нм или используют красный светофильтр). Отроят график зависимости оптической плотности растворов от концентрации Р2О5 в стандартных растворах (табл. VI.2.1, рис. VI.2.1).
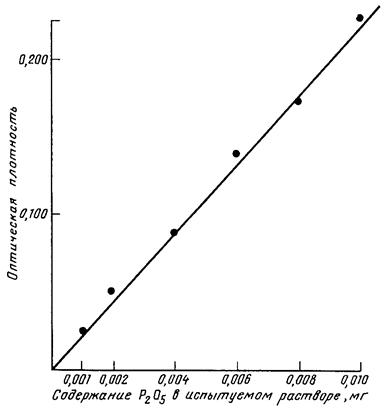
Рис. VI.2.1. Калибровочный график для определения концентрации Р2О5
Таблица VI.2.1
Результаты измерений оптической плотности стандартных растворов
|
Содержание P2O5 в испытуемом растворе, мг |
Средняя оптическая плотность |
|
|
10 |
0,001 |
0,026 |
|
0,002 |
0,049 |
|
|
0,004 |
0,087 |
|
|
0,006 |
0,138 |
|
|
0,008 |
0,172 |
|
|
0,010 |
0,227 |
VI.2.2. Приготовление рабочих растворов
0,2±0,01 М НСl. 16,4 мл концентрированной HCl разводят дистиллированной водой до 1 л.
Комплексообразователь. Кислый раствор молибденово-кислого аммония. Раствор А. 25 г молибденово-кислого аммония растворяют при нагревании в 100 мл дистиллированной воды. В остывший раствор добавляют 280 мл концентрированной H2SO4 небольшими порциями, избегая перемешивания, затем к остывшему раствору приливают 620 мл дистиллированной воды. Раствор хранят в склянке из темного стекла.
В работе используют раствор молибдата, полученный при разбавлении раствора А в 5 раз дистиллированной водой.
Восстановитель. 0,13 г SnCl2×2Н2О ч.д.а. всыпают в стеклянную пробирку, приливают сюда же 5 мл 10%-ной HCl. Пробирку погружают в термостойкий химический стакан на 100-200 мл, наполненный водой, и кипятят на электроплитке до полного растворения восстановителя.
10 % HCl. Концентрированную соляную кислоту разбавляют дистиллированной водой в три раза.
VI.2.3. Реактивы
HCl х.ч., ГОСТ 3118-67
Н2SО4 х.ч., ГОСТ 4204-66.
Аммоний молибденовокислый 4-водный х.ч., ГОСТ 3765-64.
Олово двуххлористое 2-водное (SnCl2×2Н2О) ч.д.а., ГОСТ 36-68.
Фосфорнокислый калий однозамещенный (КН2РО4) х.ч., ГОСТ 4198-65.
VI.3. Определение аммиачного азота [2-4]
Навеску свежей почвы (10-20 г) помещают в коническую колбу емкостью 500 мл. Одновременно берут 5 г почвы в бюкс для анализа на влажность.
К навеске почвы приливают 10-кратное количество 2 %-ного раствора KCl, приготовленного на безаммиачной воде. Колбу встряхивают 5 мин и оставляют на 18-20 ч. Длительное отстаивание можно заменить часовым взбалтыванием.
Контролем служит раствор KCl.
Вытяжку из первой колбы фильтруют через складчатый беззольный фильтр. Для определения количества аммиака в реакционной смеси 1-2 мл фильтрата помещают в мерную колбу на 50 мл, добавляют 10 мл дистиллированной воды, прибавляют далее 4 мл раствора фенолята натрия и сразу же 3 мл раствора гипохлорита натрия. После добавления каждого реактива смесь тщательно перемешивают. Через 20 мин объем раствора доводят дистиллированной водой до метки и интенсивность окраски просматривают на ФЭКе, используя красный светофильтр или на спектрофотометре при l=620 нм. Окраска индофенола голубого устойчива в течение 60 мин. Количество аммиачного азота находят по стандартной калибровочной кривой, используя для расчета формулу
![]()
где X - количество аммиачного азота, мг N-NH3/100 г; С - концентрация N-NH3, найденная по калибровочной кривой, мг/мл; V1 - общий объем вытяжки, мл; V2 - объем аликвотной части, мл; a - навеска почвы, г; 1000 - коэффициент для перевода мкг в мг; К - коэффициент пересчета веса почвы на вес высушенной при 100-105 °С почвы; 100 - коэффициент пересчета на 100 г почвы.
VI.3.1. Приготовление стандартных растворов сернокислого аммония и построение калибровочной кривой
0,4717 г (NH4)2SO4, высушенного до постоянного веса, помещают в мерную колбу емкостью 1000 мл, растворяют в дистиллированной воде и объем раствора доводят до метки. Концентрация исходного раствора равна 100 мкг N/мл. Для приготовления стандартных растворов в мерные колбочки емкостью 100 мл отбирают 1, 2, 5, 8, 12, 15, 18, 22, 25, 28 мл исходного раствора и доливают дистиллированной водой до метки. 1 мл каждого стандартного раствора содержит соответственно 1, 2, 5, 8, 12, 15, 18, 22 мкг N-NH3. 1 мл раствора помещают в мерную колбу емкостью 50 мл, доводят объем дистиллированной водой примерно до 10 мл, затем приливают 4 мл раствора фенолята натрия и сразу же 3 мл раствора гипохлорита натрия. После добавления каждого реактива смесь тщательно перемешивают. Через 20 мин объем смеси доводят до метки и интенсивность окраски просматривают на ФЭК с красным светофильтром, используя кювету объемом 20 мл или на спектрофотометре при длине волны 620 нм. Окраска индофенола голубого устойчива в течение 1 ч. Результаты измерений оптической плотности представлены в табл. VI.3.1, а калибровочная кривая - на рис. V.6.1. Линейная зависимость оптической плотности от концентрации N-NH3 для кюветы толщиной 20 мм сохраняется в интервале концентраций от 1 до 22 мкг /50 мл.
Таблица VI.3.1
Результаты измерений оптической плотности растворов (NH4)2SO4 на приборе ФЭК-М
|
Содержание N-NH3 в испытуемом растворе, мкг/мл |
Средняя оптическая плотность |
|
|
20 |
1 |
0,019 |
|
2 |
0,036 |
|
|
5 |
0,061 |
|
|
8 |
0,104 |
|
|
12 |
0,154 |
|
|
15 |
0,199 |
|
|
18 |
0,221 |
|
|
22 |
0,270 |
VI.3.2. Приготовление рабочих растворов
Раствор фенолята натрия. 62,5 г фенола растворяют в малом количестве эталона, добавляют 18,5 мл ацетона, этанолом доводят объем раствора до 100 мл, перемешивают (раствор А). 27 NaOH растворяют в 100 мл дистиллированной воды (раствор Б). Оба раствора хранят в холодильнике. Перед употреблением по 20 мл растворов А и Б смешивают и объем доводят до 100 мл, перемешивают.
Раствор гипохлорита натрия. Исходный препарат разбавляют водой до концентрации активного хлора, равной 0,9 %. К приготовленному исходному раствору гипохлорита натрия прибавляют дистиллированную воду в отношении 1:10. Раствор устойчивый.
VI.3.3. Реактивы
Сернокислый аммоний ((NH4)2SO4) ч.д.а., ГОСТ 3769-60.
Фенол (С6Н5ОН) ч., ГОСТ 6417-52.
Углекислый натрий безводный (Na2CO3) ч.д.а., ГОСТ 83-63.
Хлорная известь (продажная) (хлорка белая CaOCl2).
Гипохлорит натрия (натрий хлорноватистокислый NaClO).
Гидроокись натрия (NaOH) х.ч., ГОСТ 4328-66.
Раствор NaClO получают следующим образом. Тщательно размешивают 100 г хлорной извести с содержанием активного хлора 35-36 % с 170 мл дистиллированной воды в течение 15 мин и в смесь при непрерывном перемешивании вносят раствор Na2CO3 (70 г в 170 г дистиллированной воды). Масса сначала густеет, затем опять разжижается. Жидкость отделяют от осадка СаСО3 на воронке Бюхнера, используя полотняный фильтр. Получается 320 мл раствора NaClO с содержанием активного хлора 71-100 г/л.
VI.4. Определение нитратного азота дисульфофеноловым методом [2-4]
5 г свежей почвы помещают в колбу, приливают 50 мл 0,05 %-ного раствора R2SO4, приготовленного на безаммиачной дистиллированной воде, содержимое колбы взбалтывают в течение 3 мин. Одновременно берут навеску почвы для определения влажности.
Вытяжку фильтруют через складчатый фильтр из плотной бумаги, предварительно промытый горячей безаммиачной водой. Первые порции фильтрата отбрасывают, мутный раствор несколько раз перефильтровывают через тот же фильтр.
В зависимости от ожидаемого содержания нитратов берут 10-20 мл подготовленной вытяжки, помещают в фарфоровую чашечку соответствующего объема и выпаривают на электрической плитке с закрытой спиралью. После выпаривания чашки с сухим остатком охлаждают и по каплям добавляют 1 мл дисульфофеноловой кислоты.
Сухой остаток тщательно растирают с дисульфофеноловой кислотой стеклянным пестиком или стеклянной палочкой с оплавленным концом и оставляют на 10 мин. Затем в каждую чашку приливают по 15 мл дистиллированной воды, так как без добавления воды возможно разбрызгивание содержимого чашек при последующей нейтрализации кислоты щелочью. Смачивают водой всю поверхность чашек, чтобы собрать все продукты нитрования. 20 %-ным раствором NaОН или КОН кислый раствор нейтрализуют и доводят до щелочной реакции, затем добавляют избыток щелочи (одну каплю). Окрашенные в желтый цвет растворы переливают в мерные колбочки на 50 мл, обмывают чашечку вместе с палочкой 3-4 раза дистиллированной водой и прибавляют эту воду к раствору. Объем раствора доводят до метки дистиллированной водой. Растворы перемешивают и фотометрируют при l=440 нм.
Содержание нитратного азота находят по стандартной калибровочной кривой, используя для расчета формулу
![]() (VI.4.1)
(VI.4.1)
где X - количество нитратного азота, мкг N-NO3/100 г; С - концентрация N-NO3, найденная по калибровочной кривой; V1 - общий объем вытяжки (50 мл), V2 - объем аликвотной части, a - навеска почвы (5 г), К - коэффициент пересчета веса почвы на вес высушенной при температуре 100-150°С почвы.
Присутствие в вытяжке значительных количеств аммонийных солей снижает результаты определения. Для предотвращения отрицательного влияния солей перед выпариванием вытяжки в фарфоровую чашку добавляют 2-3 капли 10 %-ного раствора сернокислого калия. Присутствие в вытяжках Сl- также мешает определению. При содержании Cl более 30 мг в 1 л к 100 мл вытяжки прибавляют 0,1 г AgSO4, смесь взбалтывают 25 мин. Для осаждения избытка Аg+ в раствор добавляют в виде сухих реактивов 0,2 г Са(ОН)2 и 0,5 г MgCO3, взбалтывают 5 мин и фильтруют через складчатый фильтр. Далее выпаривают аликвоту, как указывалось выше. При удалении хлора серебром подщелачивание раствора проводят не NaOH или КОН, а аммиаком.
VI.4.1. Приготовление стандартных растворов нитрата калия и построение калибровочной кривой
Основной раствор 0,722 г KNO3, перекристаллизованного и высушенного при 100-105°С до постоянного веса, отвешивают на аналитических весах, переносят в мерную колбу на 1 л, растворяют в дистиллированной воде, объем раствора доводят водой до метки, хорошо перемешивают. 1 мл раствора содержит 0,01 мг N-NO3.
Рабочий раствор. Готовят разведением основного раствора в 50 раз водой.
Рабочий раствор содержит 0,002 мг N-NO3 в 1 мл. Его используют для приготовления образцовых стандартных растворов. Для этого 1, 5, 10, 15, 20 мл рабочего раствора выпаривают в фарфоровых чашках. После выпаривания чашки с сухим остатком охлаждают и по каплям добавляют в них 1 мл дисульфофеноловой кислоты, стараясь смочить находящийся на дне и стенках чашки остаток. Сухой остаток тщательно растирают с дисульфофеноловой кислотой стеклянным пестиком или стеклянной палочкой с оплавленным концом и оставляют на 10 мин. Затем в каждую чашку приливают по 15 мл дистиллированной воды, смачивая всю поверхность чашек, чтобы собрать все продукты нитрования. Затем 20 %-ным раствором KOH или NaOH нейтрализуют кислый раствор и доводят до щелочной реакции и добавляют избыток щелочи (1 каплю). Окрашенные в желтый цвет растворы переносят в мерные колбы на 50 мл, при этом чашку и стеклянную палочку несколько раз ополаскивают водой, которую присоединяют к раствору в мерной колбе. Объем растворов доводят водой до метки. Растворы перемешивают. Оптическую плотность растворов определяют тотчас же после развития окраски. Интенсивность окраски при стоянии испытуемых растворов снижается. Измерения проводят на приборах фотоэлектроколориметра, пользуясь синим сфетофильтром, или спектрофотометре (l=440 нм). Пример результатов измерений оптической плотности растворов на приборе ФЭК-М представлен в табл. VI.4.1, а кривая зависимости оптической плотности растворов от концентрации нитрат-иона на рис. VI.4.1.
Таблица VI.4.1
Результаты измерений оптической плотности растворов КNО3 на приборе ФЭК-М
|
Содержание N-NO3 в испытуемом растворе, мкг/мл |
Средняя оптическая плотность |
|
|
1 |
0,002 |
0,011 |
|
0,010 |
0,056 |
|
|
0,020 |
0,109 |
|
|
0,030 |
0,161 |
|
|
0,040 |
0,194 |
Линейная зависимость оптической плотности от концентрации стандартных растворов для кюветы толщиной 1 мм сохраняется в интервале концентрации от 0,1 до 2 мкг/50 мл.
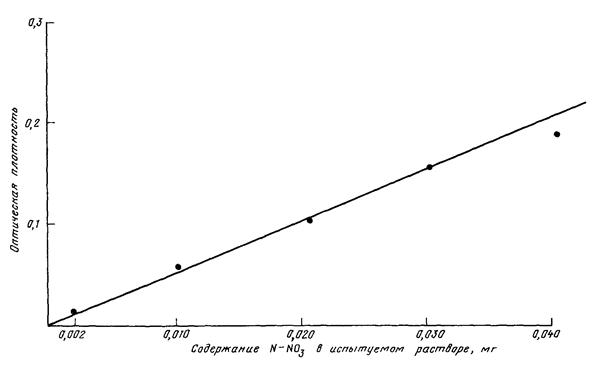
Рис. VI.4.1. Калибровочная кривая для определения концентрации N-NO3
VI.4.2. Приготовление рабочих растворов
Раствор дисульфофеноловой кислоты. Навеску 30 г чистого свежеперегнанного фенола помещают в плоскодонную колбу емкостью 500 мл, туда же приливают 200 мл концентрированной H2SO4 пл. 1.84, осторожно перемешивают, закрывают корковой пробкой со вставленной в нее длинной стеклянной трубкой, служащей обратным холодильником.
Конец трубки в пробке должен находиться на уровне ее нижней поверхности, чтобы сгущающиеся пары воды стекали по стенкам колбы и не падали в реактивную смесь. Колбу опускают в кипящую водяную баню и держат в ней 6 ч. Готовую дисульфофеноловую кислоту охлаждают и хранят в темной склянке.
20%-ный раствор NaOH или КОН. 20 г NaOH или КОН растворяют в 100 мл дистиллированной воды.
VI.4.3. Реактивы
Фенол ч., ГОСТ 6417-52.
H2SO4 пл. 1,84, ГОСТ 4204-66.
NaOH х.ч., ГОСТ 4328-66.
КОН х.ч., ГОСТ 4203-65.
Аммиак 30%, ГОСТ 3760-64.
Нитрат калия КNО3, х.ч.
AgSO4 х.ч., МРТУ 6-09-6353-69.
Са(ОН)2 ч.д.а., ГОСТ 9262-66.
МgСО3 х.ч.
VI.5. Определение сульфатов в почве [2-6]
В настоящее время существует ряд методов определения обменных сульфатов. Наиболее известны из них три: весовой, колориметрический с металлоиндикатором нитхромазо и турбидиметрический. Сравнительные характеристики методов даны в табл. VI.5.1.
Первый из перечисленных методов основан на весовом определении осадка сульфата бария, образующегося при осаждении сульфатной серы хлористым барием. Возможные потери и длительность анализа делают этот метод малопригодным.
Колориметрический метод заключается в определении сульфатов с помощью солей бария в присутствии индикатора нитхромазо. Недостатком данного метода является то, что определению сульфатов мешает присутствие катионов Са++, К+, Na+, NH3+, которые с реагентом нитхромазо дают ту же цветную реакцию.
Таблица VI.5.1
Сравнительные характеристики методов определения сульфатов в почве
|
Метол |
|||
|
весовой |
колориметрический с металлоиндикатором |
турбидиметрический |
|
|
Чувствительность, мкг |
100 |
10 |
1 |
|
Воспроизводимость, % |
80 |
20 |
90 |
|
Время анализа, ч |
4 |
0,3 |
0,5 |
В настоящих методических указаниях для определения сульфатов в почве рекомендуется турбидиметрический метод, основанный на измерении интенсивности падающего и прошедшего через мутный раствор света. Сульфат-ионы из исследуемого раствора осаждаются с помощью хлористого бария, в результате чего раствор мутнеет. Степень помутнения раствора просматривается на приборах: фотоэлектроколориметре или спектрофотометре при длине волны 365 нм. Для избежания ошибок в работе при турбидиметрическом анализе необходима стандартизация условий подготовки к турбидиметрическому анализу, так как воспроизводимость оптических свойств дисперсных систем зависит от ряда факторов, таких, как рН среды, температура, порядок смешения растворов, их концентрация, скорость смешения, время хранения образовавшейся суспензии. Данный метод является удовлетворительным при содержании сульфатов в пробе более 1 мкг и имеет ошибку измерения 10 %.
Для определения сульфатов 1-20 г (в зависимости от содержания сульфатов) воздушно-сухой почвы, а влажной - с учетом влажности, помещают в коническую колбу на 500 мл, заливают 100 мл 1 н раствора хлористого калия и взбалтывают на аппарате АВУ-1 в течение 1 ч. Затем суспензию фильтруют через плотный фильтр 2 раза для получения бесцветных прозрачных растворов. Из фильтрата берут аликвоту 2 мл и переносят ее в пробирку, затем прибавляют 2 мл 0,01 н безводного хлористого бария, рН которого на рН-метре с помощью 0,1 Н НСl доводят до 3-4 с целью установления равновесного дисперсного состава смеси, а также для устранения мешающего действия анионов, таких как F-, МnO4-, СO3-- и др. Спустя 1 ч степень помутнения растворов просматривают на спектрофотометре при длине волны 365 нм в кюветах с толщиной слоя 1 см по сравнению с контролем. Для приготовления контрольного раствора, не содержащего ионы SO4--, 2 мл бидистиллированной воды смешивают с 2 мл раствора хлористого бария (табл. VI.5.2). В случае высокого содержания сульфатов перед взятием аликвоты раствор разводят в несколько раз, что затем учитывают в расчетах.
Таблица VI.5.2
Схема приготовления стандартных растворов
Содержание ионов SO4-- ведется по формуле
![]()
где X - концентрация ионов SO4--, г/кг, в почве; С - содержание ионов SO4-- в растворе, найденная по калибровочной кривой, мкг; V1 - общий объем вытяжки, 100 мл; V2 - аликвота, 2 мл; а - навеска почвы, 1 г.
Численные коэффициенты даны для перевода единицы измерения содержания ионов SO4-- из мкг/г в г/кг.
VI.5.1. Приготовление стандартных растворов и построение калибровочной кривой
Для приготовления основного стандартного раствора на аналитических весах отвешивают 54,42 мг K2SO4 марки "ОСЧ" и растворяют в 1 л бидистиллированной воды.
Получают раствор, 1 мл которого содержит 30 мкг SO4--. Из полученного раствора готовят серию рабочих стандартных растворов (табл. VI.5.2), содержащих 0, 6, 15, 30, 45, 60 мкг SO4--. Для этого пипеткой на 2 мл отбирают соответственно 0, 0,2, 0,5, 1,0, 1,5, 20 мл исходного стандартного раствора и переносят в пробирки, затем в каждую пробирку прибавляют соответственно 2, 1,8, 1,5, 1,0, 0,5, 0 мл бидистиллированной воды и тщательно перемешивают. Затем прибавляют по 2 мл 0,01 М раствора ВаСl2 и тут же тщательно перемешивают. Оптическую плотность D полученных растворов определяют на спектрофотометре и строят калибровочный график зависимости D от концентрации сульфат-ионов в стандартном растворе (табл. VI.5.3. рис. VI.5.1).
Таблица VI.5.3
Результаты измерения оптической плотности растворов K2SO4 на СЭФ-16
|
Содержание сульфат-иона в испытуемом растворе, мкг |
Оптическая плотность |
|
|
1 см |
6 |
0,039 |
|
15 |
0,070 |
|
|
30 |
0,123 |
|
|
45 |
0,153 |
|
|
60 |
0,185 |
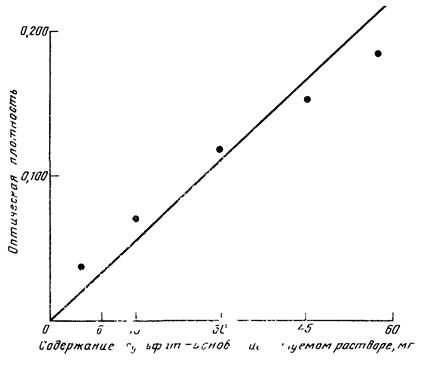
Рис. VI.5.1. Калибровочная кривая для определения концентрации SO4--
VI.5.2. Приготовление рабочих растворов
1 н раствор хлористого калия. 75,7 г растворяют в 1 л дистиллированной воды.
Раствор сернокислого калия. 54,42 мг K2SO4 марки "ОСЧ" растворяют в 1 л дистиллированной воды.
0,01 М раствор хлористого бария. 2,08 г ВaСl2 растворяют в 1 литре дистиллированной воды.
0,1 н раствор HCl. Готовится из фикоанала.
VI.5.3. Реактивы
Калий хлористый (KCl) х.ч., ГОСТ 4234-69.
Барий хлористый (ВaСl2) х.ч., МРТУ 6-09-4505-68.
Калий сернокислый (К2SO4) х.ч., ГОСТ 4145-65.
Фиксанал соляной кислоты 0,1 н.
Список литературы
3. Агрохимические методы исследования почв. - М.: Наука, 1975.- 655 с.
4. Аринушкина Е.В. Руководство по химическому анализу почв. -М., Изд-во МГУ, 1961.- 490 с.
5. Айдинян Р.Х., Иванова М.С., Соловьева Т.Г. Методы извлечения и определения различных форм серы в почвах и растениях. М., 1968.
6. Руководство по методам определения вредных веществ в атмосферном воздухе. - М.: Медицина, 1974.- 51 с.
СОДЕРЖАНИЕ