Государственное санитарно-эпидемиологическое
нормирование
Российской Федерации
Федеральная служба по надзору в сфере защиты прав
потребителей
и благополучия человека
4.1. МЕТОДЫ КОНТРОЛЯ. ХИМИЧЕСКИЕ ФАКТОРЫ
Определение остаточных количеств
действующих веществ пестицидов
в зеленой массе, зерне, масле, семенах
Сборник методических указаний
по методам контроля
МУК 4.1.2988-12; 4.1.2994-12;
4.1.3002-12
Москва • 2013
1. Рекомендованы к утверждению Комиссией по государственному санитарно-эпидемиологическому нормированию при Федеральной службе по надзору в сфере защиты прав потребителей и благополучия человека (протокол от 22.12.2011 № 2).
2. Утверждены Руководителем Федеральной службы по надзору в сфере защиты прав потребителей и благополучия человека. Главным государственным санитарным врачом Российской Федерации Г.Г. Онищенко 19 марта 2012 г.
3. Введены в действие с момента утверждения.
4. Введены впервые.
СОДЕРЖАНИЕ
|
УТВЕРЖДАЮ Руководитель Федеральной службы по надзору в сфере защиты прав потребителей и благополучия человека, Главный государственный санитарный врач Российской Федерации Г.Г. Онищенко 19 марта 2012 г. Дата введения: с момента утверждения |
4.1. МЕТОДЫ КОНТРОЛЯ. ХИМИЧЕСКИЕ ФАКТОРЫ
Определение остаточных количеств флуроксипира
в зеленой массе растений, зерне и масле кукурузы
методом капиллярной газожидкостной хроматографии
Методические указания
МУК 4.1.2988-12
Свидетельство об аттестации от 25.07.2011 № 01.5.04.014/01.00043/2011.
Настоящие методические указания устанавливают метод капиллярной газожидкостной хроматографии для определения в зеленой массе растений, зерне и масле кукурузы массовой концентрации флуроксипира в диапазоне концентраций 0,01 - 0,08 мг/кг.
Название действующего вещества по номенклатуре ICO: флуроксипир.
Название по номенклатуре IUPAC: 4-амино-3,5-дихлор-6-фтор-2-пиридилоксиуксусная кислота.
Структурная формула:
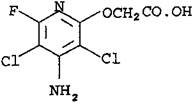
Брутто формула: C7H5Cl2FN2O3.
Молекулярная масса: 255,0.
Белое кристаллическое вещество, температура плавления - 232 - 233 °C. Давление пара (25 °C) 0,126 мПа (9,42 · 10-7 мм рт. ст.). Растворимость (20 °C, в г/л): в воде - 0,091, метаноле - 43,7, ацетоне - 64,7, этилацетате - 11,8, ксилоле - 0,3.
ЛД50 (в мг/кг) для крыс > 5000. СК50 (в мг/л): для радужной форели и серебряного карпа - 0,7 (96 ч); для дафний > 0,5 (48 ч). ЛД50 для пчел > 0,1 мг/особь.
Гигиенические нормативы: ПДК в воде водоемов - 0,01 мг/дм3, ОДК в почве - 0,2 мг/кг, МДУ в зерне хлебных злаков, луке - 0,05 мг/кг.
Область применения: гербицид системного ауксиноподобного действия для послевсходовой некорневой обработки против широкого круга широколистных сорных растений (включая подмаренник цепкий и мокрицу-звездчатку) в зерновых, декоративных культурах; против многолетних сорных растений в плодовых садах (включая щавель, вьюнок и другие).
1. Метрологическая характеристика метода
При соблюдении всех регламентированных условий проведения анализа в точном соответствии с данной методикой погрешность (и ее составляющие) результатов измерений при доверительной вероятности P = 0,95 не превышает значений (табл. 1) для соответствующих диапазонов концентраций.
Таблица 1
|
Показатель повторяемости (относительное среднеквадратическое отклонение повторяемости), sr, % |
Показатель промежуточной прецизионности (относительное среднеквадратическое отклонение в условиях вариации факторов «время», «оператор» в одной лаборатории), sRt, % |
Показатель воспроизводимости (относительное среднеквадратическое отклонение воспроизводимости), sr, % |
Показатель точности* (границы относительной погрешности при вероятности P = 0,95), ± δ, % |
|
|
Зеленая масса |
8 |
9 |
11 |
22 |
|
от 0,01 до 0,08 вкл. |
||||
|
Зерно |
7 |
8 |
10 |
20 |
|
от 0,01 до 0,08 вкл. |
||||
|
Масло |
9 |
11 |
13 |
24 |
|
от 0,01 до 0,08 вкл. |
||||
|
* Соответствует расширенной неопределенности Uотн при коэффициенте охвата k = 2 |
||||
Таблица 2
Полнота извлечения вещества, стандартное отклонение, доверительный интервал среднего результата для флуроксипира (n = 20, Р = 0,95)
|
Объект анализа |
Предел обнаружения, мг/кг |
Диапазон определяемых концентраций, мг/кг |
Среднее значение определения, % |
Стандартное отклонение, S, % |
Доверительный интервал среднего результата, ±, % |
|
Зеленая масса |
0,01 |
0,01 - 0,08 |
83,2 |
7,0 |
3,3 |
|
Зерно |
0,01 |
0,01 - 0,08 |
81,7 |
5,2 |
2,5 |
|
Масло |
0,01 |
0,01 - 0,08 |
78,6 |
8,3 |
3,9 |
2. Метод измерения
Метод основан на извлечении остаточных количеств флуроксипира из анализируемого объекта органическими растворителями, проведении очистки экстракта перераспределением в системе несмешивающихся растворителей и метилировании флуроксипира диазометаном. Количественное определение проводят методом абсолютной калибровки с применением капиллярной газожидкостной хроматографии и использованием детектора электронного захвата.
Метод специфичен в присутствии других применяемых пестицидов. Проведение очистки экстрактов, а также использование капиллярной колонки и селективного детектора позволяет устранять влияние коэкстрактивных веществ на результаты анализа.
3. Средства измерения, реактивы, вспомогательные устройства и материалы
3.1. Средства измерения
|
Газовый хроматограф с детектором электронного захвата и хроматографической кварцевой капиллярной колонкой длиной 15 м, внутренним диаметром 0,32 мм с неподвижной фазой SE-52, толщина пленки 0,4 мкм |
|
|
Весы аналитические типа ВЛА-200 |
ГОСТ 24104-01 |
|
Весы лабораторные типа ВЛКТ-500 |
ГОСТ 24104-80 |
|
Колбы-концентраторы объемом 250 см3 |
|
|
Колбы плоскодонные объемом 100 и 300 см3 |
|
|
Колбы мерные со шлифом объемом 25, 50, 100 см3 |
|
|
Микрошприц МШ-10 |
ТУ 2-833-106 |
|
Пипетки градуированные объемом 1, 2, 5 и 10 см3 |
|
|
Пробирки мерные со шлифом объемом 5,0 см3 |
|
|
Стаканы химические объемом 100, 200 и 500 см3 |
|
|
Флаконы стеклянные (типа пенициллиновых) объемом 2,0, 3,0 и 5,0 см3 |
ТУ 64-2-10-87 |
|
Цилиндры мерные объемом 25 и 250 см3 |
Примечание: Допускается использование средств измерения с аналогичными характеристиками.
3.2. Реактивы
|
Аналитический стандарт флуроксипира |
|
|
Азот газообразный высокой чистоты |
ТУ 301-07-25-89 |
|
Ацетон, осч |
ТУ 2633-004-11291058-94 |
|
Ацетонитрил для хроматографии, хч |
ТУ 6-09-4326-76 |
|
Вода дистиллированная |
ГОСТ 7602-72 |
|
н-Гексан, хч |
ТУ 6-09-3375-78 |
|
Дихлорметан, хч |
ТУ 6-09-2662-77 |
|
Изооктан эталонный |
|
|
Калия гидроокись, чда |
|
|
Натрий серно-кислый б/в (сульфат), чда |
|
|
Натрий хлористый, чда |
|
|
N-Нитрозометилмочевина, хч |
ТУ 6-09-11-1643-82 |
|
Серная кислота, осч |
|
|
Смесь н-гексан:диэтиловый эфир, 50, 50, по объему |
|
|
Эфир диэтиловый, чда |
ТУ 2600-001-43852015-05 |
Примечание: Допускается использование реактивов с аналогичной квалификацией.
3.3. Вспомогательные устройства и материалы
|
Аппарат для встряхивания |
ТУ 64-1-1081-73 |
|
Ванна ультразвуковая УЗВ/100 ТН |
|
|
Вата медицинская |
ТУ 9393-001-00302238-97 |
|
Воронки делительные объемом 250 и 500 см3 |
|
|
Воронки химические конусные |
|
|
Индикаторная бумага универсальная |
ТУ 6-09-1181-76 |
|
Колпачки алюминиевые для герметизации флаконов |
|
|
Мельница электрическая лабораторная |
ТУ 46-22-236-79 |
|
Насос водоструйный |
ГОСТ 10696-75 |
|
Ротационный вакуумный испаритель LABOROTA 4000 |
|
|
Приспособление для обжима колпачков на флаконах |
ТУ 42-2-2442-73 |
|
Установка для перегонки растворителей при атмосферном давлении |
|
|
Установка для упаривания растворителей в токе азота |
|
|
Фильтры бумажные «красная лента» |
ТУ 2642-001-42624157-98 |
|
Фильтры бумажные «белая лента» |
ТУ 2642-001-42624157-98 |
|
Фильтры бумажные «синяя лента» |
ТУ 2642-001-42624157-98 |
|
Электроплитка |
Примечание: Допускается использование другого оборудования с аналогичными техническими характеристиками.
4. Требования безопасности
4.1. При проведении работы необходимо соблюдать правила техники безопасности, установленные для работ с токсичными, едкими, легковоспламеняющимися веществами (ГОСТ 12.1.005, 12.1.007). Организация обучения работников по безопасности труда (ГОСТ 12.0.004).
При выполнении измерений с использованием газового хроматографа и работе с электроустановками соблюдать правила электробезопасности в соответствии с требованиями ГОСТ 12.1.019 и инструкциями по эксплуатации приборов.
4.2. Помещение лаборатории должно быть оборудовано приточно-вытяжной вентиляцией, соответствовать требованиям пожарной безопасности (ГОСТ 12.1.004) и иметь средства пожаротушения (ГОСТ 12.4.009). Содержание вредных веществ в воздухе лабораторного помещения не должно превышать норм, установленных ГН 2.2.5.1313-03 «Предельно допустимые концентрации (ПДК) вредных веществ в воздухе рабочей зоны».
5. Требования к квалификации операторов
Измерения может выполнять специалист-химик, имеющий опыт работы методом капиллярной газожидкостной хроматографии, ознакомленный с руководством по эксплуатации газового хроматографа, освоивший данный метод и подтвердивший экспериментально соответствие получаемых результатов нормативам контроля погрешности измерений по п. 13.
6. Условия измерения
При выполнении измерения соблюдают следующие условия:
• процессы приготовления растворов и подготовки проб к анализу проводят при температуре воздуха лабораторного помещения (20 ± 5) °C и относительной влажности воздуха не более 80 %;
• выполнение измерения на газовом хроматографе проводят в условиях, рекомендованных технической документацией к прибору.
7. Отбор проб и хранение
Отбор проб для анализа проводят в соответствии с «Унифицированными правилами отбора проб сельскохозяйственной продукции, продуктов питания и объектов окружающей среды для определения микроколичеств пестицидов» от 21.08.1979 № 2051-79.
8. Подготовка к определению
8.1. Кондиционирование колонки
Капиллярную хроматографическую колонку устанавливают в газовый хроматограф и перед анализом кондиционируют при температуре 280 °C до установления нулевой линии.
8.2. Подготовка и очистка растворителей
Перед началом работы проверяют чистоту применяемых органических растворителей. Для этого 100 см3 растворителя упаривают в ротационном вакуумном испарителе при температуре 40 °C до объема 1,0 см3 и хроматографируют. При обнаружении мешающих определению примесей очистку растворителей производят в соответствии с общепринятыми методиками.
8.3. Приготовление эфирного раствора диазометана (из расчета метилирования экстрактов 2 проб)
N-Нитрозометилмочевину массой (0,5 ± 0,01) г помещают во флакон объемом 2,0 - 3,0 см3 и герметизируют резиновой пробкой и колпачком с помощью приспособления для обжима колпачков на флаконах. В другой флакон объемом 5,0 см3 вносят диэтиловый эфир объемом 4,0 см3, герметизируют резиновой пробкой и колпачком и охлаждают в морозильной камере холодильника в течение 30 мин.
После этого флаконы через предварительно проколотые пробки соединяют гибкой тефлоновой трубкой (внутр. диам. ~ 1,5 - 2,0 мм), одним концом погружая ее в диэтиловый эфир на всю глубину (флакон с охлажденным диэтиловым эфиром обязательно должен еще иметь свободный выход в атмосферу). Во флакон с нитрозометилмочевиной, используя шприц с тонкой иглой и прокалывая пробку, добавляют по каплям по стенке 50 % водный раствор гидроокиси калия (~ 0,3 см3) до прекращения реакции. Диэтиловый эфир при насыщении диазометаном окрашивается в ярко желтый цвет.
Внимание! Приготовление эфирного раствора диазометана и процедуру метилирования проводят в работающем вытяжном шкафу.
8.4. Приготовление градуировочных растворов
Основной раствор флуроксипира с содержанием 100 мкг/см3 готовят растворением в ацетоне 0,01 г аналитического стандарта флуроксипира в мерной колбе объемом 100 см3. Раствор хранят в холодильнике при температуре 4 - 6 °C не более трех месяцев.
Рабочие стандартные растворы с концентрациями 0,8, 0,4, 0,2 и 0,1 мкг/см3 готовят из основного стандартного раствора флуроксипира последовательным разбавлением ацетоном. Рабочие растворы хранят в холодильнике при температуре 4 - 6 °C не более месяца. В модельных опытах при изучении полноты извлечения флуроксипира используют ацетоновые растворы стандартного вещества.
Для приготовления градуировочных растворов в мерные пробирки со шлифом объемом 5,0 см3 вносят по 1,0 см3 рабочих растворов флуроксипира с концентрациями 0,1, 0,2, 0,4 и 0,8 мкг/см3. Растворитель в пробирках упаривают в токе азота досуха и проводят метилирование флуроксипира по п. 8.4.1.
8.4.1. Метилирование флуроксипира
В пробирки с сухим остатком добавляют по 2,0 см3 свежеприготовленного по п. 8.3 эфирного раствора диазометана. Пробирки закрывают пробками и ставят на 12 - 14 ч (на ночь) в холодильник с температурой 4 - 6 °C. После этого диэтиловый эфир в пробирках упаривают в токе азота досуха и сухой остаток растворяют в 2,0 см3 изооктана.
8.5. Построение градуировочной характеристики
Для построения градуировочного графика в инжектор хроматографа (п. 9.3) вводят по 1 мм3 приготовленных по пп. 8.4 и 8.4.1 растворов, содержащих флуроксипир (в виде производного) в концентрациях 0,1, 0,2, 0,4 и 0,8 мкг/см3. Осуществляют не менее трех параллельных измерений и находят среднее значение высоты (площади) хроматографического пика для каждой концентрации. Строят градуировочный график зависимости высоты (площади) хроматографического пика в мм (мм2) от концентрации флуроксипира в градуировочном растворе (мкг/см3). Градуировочную характеристику необходимо проверять при замене реактивов, хроматографической колонки или элементов хроматографической системы, изменении условий хроматографирования, а также при отрицательном результате контроля градуировочного коэффициента.
Градуировочную зависимость признают стабильной при выполнении следующего условия:
![]() где
где
C - аттестованное значение массовой концентрации флуроксипира в градуировочном растворе,
Cк - результат контрольного измерения массовой концентрации флуроксипира в градуировочном растворе,
λконтр. - норматив контроля градуировочного коэффициента, % (λконтр = 10 % при P = 0,95).
8.6. Первичная обработка проб
Пробы растений кукурузы измельчают, зеленую массу перемешивают и выделяют аналитические пробы. Для длительного хранения аналитические пробы зеленой массы растений помещают в морозильную камеру с температурой -18 °C и хранят в закрытой стеклянной или полиэтиленовой таре.
Пробы зерна перед анализом рассыпают на бумаге или кальке и пинцетом удаляют включения. Зерно измельчают на лабораторной мельнице и после перемешивания измельченной массы готовят усредненные аналитические пробы.
Для исследовательских целей допускается получение в лаборатории масла из проб измельченного зерна методом экстракции органическими растворителями при температуре не выше 40 °C. Пробы масла хранят при 4 - 6 °C в закрытой стеклянной таре не более 30 сут.
9. Проведение определения
9.1. Определение флуроксипира в зеленой массе и зерне кукурузы
Аналитическую пробу зеленой массы или зерна массой (10,0 ± 0,1) г помещают в плоскодонную колбу объемом 300 см3, добавляют 150 см3 ацетона, слегка встряхивают и подвергают обработке в ультразвуковой ванне в течение 10 мин при комнатной температуре. После этого содержимое колбы фильтруют через бумажный фильтр «красная лента» в колбу-концентратор объемом 250 см3. Содержимое колбы с пробой промывают 50 см3 ацетона и также фильтруют в колбу-концентратор.
При использовании аппарата для встряхивания в плоскодонную колбу с аналитической пробой вносят 150 см3 ацетона и встряхивают в течение 60 мин. После этого содержимое колбы фильтруют через бумажный фильтр «красная лента» в колбу-концентратор объемом 250 см3. Содержимое колбы с пробой промывают 50 см3 ацетона и также фильтруют в колбу-концентратор.
Колбу-концентратор с объединенным экстрактом подсоединяют к ротационному вакуумному испарителю и упаривают растворитель до объема 10 - 20 см3 при температуре 40 °C. В колбу-концентратор добавляют 200 см3 дистиллированной воды, 2,0 см3. 5,0 %-го водного раствора серной кислоты и содержимое колбы перемешивают встряхиванием. Колбу-концентратор помещают в холодильник и выдерживают при температуре 4 - 6 °C в течение 4 - 5 ч. После этого содержимое колбы фильтруют через бумажный фильтр «белая лента» в делительную воронку объемом 500 см3. В воронку добавляют 10 %-й водный раствор гидроокиси калия до pH 9 - 10, 30 см3 насыщенного водного раствора хлористого натрия и после перемешивания 75 см3 дихлорметана. Содержимое воронки энергично встряхивают в течение 2 мин. После 15-минутного отстаивания нижний дихлорметановый слой сливают и отбрасывают. Процедуру очистки экстракта повторяют с использованием 50 см3 дихлорметана. Далее в воронку добавляют 40 см3 насыщенного водного раствора хлористого натрия и после перемешивания 75 см3 н-гексана. Содержимое воронки энергично встряхивают в течение 2 мин. После 5-минутного отстаивания нижний ацетоно-водный слой сливают в химический стакан объемом 500 см3, а верхний гексановый слой сливают и отбрасывают.
Водный раствор пробы, находящийся в химическом стакане, подкисляют концентрированной серной кислотой до pH 2,0 и переносят в чистую делительную воронку объемом 500 см3. В воронку добавляют 75 см3 смеси н-гексан-диэтиловый эфир (50:50) и встряхивают в течение 2 мин. После полного разделения слоев нижний водный слой сливают в химический стакан, а верхний гексано-эфирный слой фильтруют через фильтр «синяя лента» со слоем безводного сульфата натрия (толщина слоя ~ 1,0 - 1,5 см) в колбу-концентратор объемом 250 см3. Экстрагирование и фильтрование повторяют с использованием 50 см3 смеси н-гексан-диэтиловый эфир (50:50). Нижний водный слой отбрасывают.
Колбу-концентратор с объединенным гексано-эфирным экстрактом подсоединяют к ротационному вакуумному испарителю и упаривают растворители при температуре 40 °C до объема 3 - 5 см3. Остаток экстракта количественно переносят в мерную пробирку со шлифом объемом 5,0 см3 и упаривают растворители в токе азота досуха при температуре 40 °C. Метилирование флуроксипира проводят по п. 8.4.1, а газохроматографический анализ - по п. 9.3.
9.2. Определение флуроксипира в масле кукурузы
Аналитическую пробу масла массой (10,0 ± 0,1) г растворяют в 50 см3 н-гексана (насыщенного ацетонитрилом) в плоскодонной колбе объемом 100 см3 и после этого гексановый раствор масла переносят в делительную воронку объемом 250 см3. Колбу промывают 50 см3 ацетонитрила (насыщенного н-гексаном) и переносят его в воронку. Содержимое воронки встряхивают в течение 2 мин. После 5-минутного отстаивания нижний ацетонитрильный слой сливают в колбу-концентратор объемом 250 см3. Плоскодонную колбу промывают 25 см3 ацетонитрила (насыщенного н-гексаном) и также переносят в делительную воронку (250 см3). Содержимое воронки встряхивают в течение 2 мин, отстаивают 5 мин и нижний ацетонитрильный слой объединяют в колбе-концентраторе с предыдущим. Верхний гексановый слой отбрасывают.
Колбу-концентратор с объединенным ацетонитрильным экстрактом подсоединяют к ротационному вакуумному испарителю и упаривают растворитель досуха при температуре 50 °C. Сухой остаток растворяют в 20 см3 ацетона. К раствору добавляют 200 см3 дистиллированной воды, 2,0 см3 5,0 %-го водного раствора серной кислоты и содержимое колбы перемешивают встряхиванием. Колбу-концентратор помещают в холодильник и выдерживают при температуре 4 - 6 °C в течение 4 - 5 ч. После этого содержимое колбы фильтруют через бумажный фильтр «белая лента» в делительную воронку объемом 500 см3. В воронку добавляют 10 %-й водный раствор гидроокиси калия до pH 9 - 10, 30 см3 насыщенного водного раствора хлористого натрия и, после перемешивания, 75 см3 дихлорметана. Содержимое воронки энергично встряхивают в течение 2 мин. После 15-минутного отстаивания нижний дихлорметановый слой сливают и отбрасывают. Процедуру очистки экстракта повторяют с использованием 50 см3 дихлорметана. Далее в воронку добавляют 40 см3 насыщенного водного раствора хлористого натрия и, после перемешивания, 75 см3 н-гексана. Содержимое воронки энергично встряхивают в течение 2 мин. После 5-минутного отстаивания нижний ацетоно-водный слой сливают в химический стакан объемом 500 см3, а верхний гексановый слой сливают и отбрасывают.
Водный раствор пробы, находящийся в химическом стакане, подкисляют концентрированной серной кислотой до pH 2,0 и переносят в чистую делительную воронку объемом 500 см3. В воронку добавляют 75 см3 смеси н-гексан-диэтиловый эфир (50:50) и встряхивают в течение 2 мин. После полного разделения слоев нижний водный слой сливают в химический стакан, а верхний гексано-эфирный слой фильтруют через фильтр «синяя лента» со слоем безводного сульфата натрия (толщина слоя ~ 1,0 - 1,5 см) в колбу-концентратор объемом 250 см3. Экстрагирование и фильтрование повторяют с использованием 50 см3 смеси н-гексан-диэтиловый эфир (50:50). Нижний водный слой отбрасывают.
Колбу-концентратор с объединенным гексано-эфирным экстрактом подсоединяют к ротационному вакуумному испарителю и упаривают растворители при температуре 40 °C до объема 3 - 5 см3. Остаток экстракта количественно переносят в мерную пробирку со шлифом объемом 5,0 см3 и упаривают растворители в токе азота досуха при температуре 40 °C. Метилирование флуроксипира проводят по п. 8.4.1, а газо-хроматографический анализ - по п. 9.3.
9.3. Условия хроматографирования
Газовый хроматограф с детектором электронного захвата и хроматографической кварцевой капиллярной колонкой длиной 15 м, внутренним диаметром 0,32 мм с неподвижной фазой SE-52, толщина пленки 0,4 мкм.
Температура колонки: программирование от 120 (1 мин) до 280 °C (20 мин) со скоростью 8,0 °C/мин. Температура испарителя - 250 °C, детектора - 300 °C. Расход газов: газа-носителя (азот в/ч) - 1,5 см3/мин, дополнительного газа (азот в/ч) к детектору - 40 см3/мин. Пробы вводят в инжектор хроматографа в режиме разделения потока газа-носителя 1:10. Количество аликвоты, вводимое в хроматограф - 1 мкл. Время удерживания флуроксипира (в виде производного): (13,8 ± 0,03) мин.
10. Обработка результатов анализа
Количественное определение флуроксипира проводят методом абсолютной калибровки и вычисляют по формуле:
![]() где
где
X - содержание флуроксипира в пробе, мг/кг;
H2 - высота (площадь) пика анализируемого вещества, мм (мм2);
H1 - высота (площадь) пика стандартного вещества, мм (мм2);
C - концентрация стандартного раствора флуроксипира, мкг/см3;
V - объем экстракта, подготовленного для хроматографирования, см3;
P - масса (г) аналитической пробы.
Содержание остаточных количеств флуроксипира в анализируемом образце вычисляют как среднее из двух параллельных определений. При получении зашкаленных пиков анализируемый экстракт разбавляют изооктаном.
11. Проверка приемлемости результатов параллельных определений
За результат анализа принимают среднее арифметическое результатов двух параллельных определений, расхождение между которыми не превышает предела повторяемости (1):
![]() где (1)
где (1)
X1, X2 - результаты параллельных определений, мг/кг;
r - значение предела повторяемости (r = 2,8sr).
При невыполнении условия (1) выясняют причины превышения предела повторяемости, устраняют их и вновь выполняют анализ.
12. Оформление результатов
Результат анализа представляют в виде:
![]() мг/кг
при вероятности P = 0,95, где
мг/кг
при вероятности P = 0,95, где
![]() - среднее
арифметическое результатов определений, признанных приемлемыми, мг/кг;
- среднее
арифметическое результатов определений, признанных приемлемыми, мг/кг;
D - граница абсолютной погрешности, мг/кг;
![]() где
где
δ - граница относительной погрешности методики (показатель точности в соответствии с диапазоном концентраций), %.
В случае, если содержание компонента менее нижней границы диапазона определяемых концентраций, результат анализа представляют в виде:
«содержание вещества в пробе менее 0,01 мг/кг», где: 0,01 мг/кг - предел обнаружения флуроксипира в анализируемых объектах.
13. Контроль качества результатов измерений
Оперативный контроль погрешности и воспроизводимости измерений осуществляется в соответствии с ГОСТ Р ИСО 5725-1-6-02 «Точность (правильность и прецизионность) методов и результатов измерений».
Стабильность результатов измерений контролируют перед проведением измерений, анализируя один из градуировочных растворов. Плановый внутрилабораторный оперативный контроль процедуры выполнения анализа проводится с применением метода добавок.
Величина добавки Cp должна удовлетворять условию:
Cp = Dл,X + Dл,X', где
± Dл,X (± Dл,X') - характеристика погрешности (абсолютная погрешность) результатов анализа, соответствующая содержанию компонента в испытуемом образце (расчетному значению содержания компонента в образце с добавкой, соответственно), мг/кг; при этом:
Dл = ± 0,84D, где
D - граница абсолютной погрешности, мг/кг:
![]() где
где
δ - граница относительной погрешности методики (показатель точности в соответствии с диапазоном концентраций), %.
Результат контроля процедуры Kк рассчитывают по формуле:
Kк = X' - X - Cp, где
X', X, Cp - среднее арифметическое результатов параллельных определений (признанных приемлемыми) содержания компонента в образце с добавкой, испытуемом образце, концентрация добавки, соответственно, мг/кг.
Норматив контроля K рассчитывают по формуле:
![]()
Проводят сопоставление результата контроля процедуры (Kк) с нормативом контроля (K).
Если результат контроля процедуры удовлетворяет условию
процедуру анализа признают удовлетворительной. При невыполнении условия (2) процедуру контроля повторяют. При повторном невыполнении условия (2) выясняют причины, приводящие к неудовлетворительным результатам, и принимают меры к их устранению.
Расхождение между результатами измерений, выполненных в двух разных лабораториях, не должно превышать предела воспроизводимости (R):
![]() где (3)
где (3)
X1, X2 - результаты измерений в двух разных лабораториях, мг/кг;
R - предел воспроизводимости (в соответствии с диапазоном концентраций), %.
14. Разработчики
Долженко В.И., Тарарин П.А., Маханькова Т.А., Редюк С.И., Бурлакова Ю.В. (ГНУ ВИЗР Россельхозакадемии).
Методика прошла метрологическую экспертизу (Свидетельство об аттестации № 01.5.04.014/01.00043/2011) и внесена в Федеральный реестр (ФР. 1.31.2011.10795).