Государственное санитарно-эпидемиологическое
нормирование
Российской Федерации
4.1. МЕТОДЫ КОНТРОЛЯ. ХИМИЧЕСКИЕ ФАКТОРЫ
Определение
остаточных количеств
пестицидов в сельскохозяйственном
сырье, пищевых продуктах и
объектах окружающей среды
Сборник
методических указаний
по методам контроля
МУК
4.1.2907-11; 4.1.2923
- 4.1.2925-11;
4.1.2938-11
Москва • 2011
1. Разработаны сотрудниками ГНУ Всероссийского НИИ защиты растений Россельхозакадемии.
2. Рекомендованы к утверждению Комиссией по санитарно-эпидемиологическому нормированию Федеральной службы по надзору в сфере защиты прав потребителей и благополучия человека (протокол от 2.06.2011 № 1).
3. Утверждены Руководителем Федеральной службы по надзору в сфере защиты прав потребителей и благополучия человека, Главным государственным санитарным врачом Российской Федерации Г.Г. Онищенко 12 июля 2011 г.
4. Введены в действие с момента утверждения.
5. Введены впервые.
СОДЕРЖАНИЕ
|
|
УТВЕРЖДАЮ Руководитель
Федеральной службы Г.Г. Онищенко 12 июля 2011 г. Дата введения: с момента утверждения |
4.1. МЕТОДЫ КОНТРОЛЯ. ХИМИЧЕСКИЕ ФАКТОРЫ
Определение остаточных количеств бифентрина
в капусте, зерне гороха, сои и соевом масле
методом газожидкостной хроматографии
Методические указания
МУК 4.1.2938-11
Настоящие методические указания устанавливают метод газожидкостной хроматографии для определения в капусте, зерне гороха, сои и соевом масле массовой концентрации бифентрина в диапазоне концентраций 0,01 - 0,1 мг/кг.
Бифентрин (ISO).
Структурная формула:
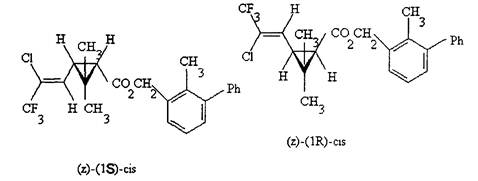
СА: (2-метил[1,1'-дифенил]-3-ил)метил 3-(2-хлор-3,3,3-трифтор-1-пропенил)-2,2-диметилциклопропанкарбоксилат
Мол. масса: 422,9.
Брутто формула: C23H22ClF3O2.
Химически чистый препарат - очень вязкая маслообразная жидкость, имеющая тенденцию к затвердеванию.
Температура плавления: 68 - 70,6 °С
Давление паров при 25 °С 0,024 mРа
Коэффициент распределения н-октанол/вода: Kow log Р > 6
Растворимость в воде < 1μг/л;
Хорошо растворим в ацетоне (125 г/100 мл), хлороформе, дихлорметане, диэтиловом эфире и толуоле; слабо растворим в гептане и метаноле.
Устойчив в водном растворе при рН 5 - 9 (21 °С) в течение 21 дня.
Стабилен в течение 2 лет при температуре от 25 до 50 °С. В почве период полураспада DT50 65 - 125 дней. Термостабилен до 250 °С; фотостабильность: естественный свет - DT50 = 255, искусственная лампа - DT50 = 11,9 дн.
Класс токсичности по ВОЗ - II. Оральная токсичность (LD50) для крыс 54,5 мг/кг
Гигиенические нормативы: ВМДУ бифентрина в капусте 1,0 мг/кг, для гороха и сои не установлены.
Область применения: инсектицид широкого спектра действия для борьбы с тлей, клещом, щитовкой, белокрылкой и другими на фруктовых, овощных, бахчевых, бобовых и зерновых культурах.
2. Методика определения бифентрина в капусте, зерне гороха, сои и соевом масле методом газожидкостной хроматографии
2.1. Принцип метода
Метод определения бифентрина в растительных объектах основан на экстракции пестицида органическим растворителем и очистке перераспределением между двумя несмешивающимися растворителями и, при необходимости, на колонке с силикагелем. Количественное определение бифентрина в капусте и горохе проводят методом газожидкостной хроматографии на насадочной колонке, в зерне и масле сои - на капиллярной колонке с использованием электронозахватного детектора (ДПР).
Идентификация бифентрина проводится по времени удерживания, количественное определение - методом абсолютной калибровки.
2.2. Метрологическая характеристика метода
Метрологическая характеристика метода представлена в табл. 1 и 2.
Метрологическая характеристика метода
|
Диапазон измерений, массовая концентрация, мг/кг |
Показатель повторяемости (относительное среднеквадратическое отклонение повторяемости), σr, % |
Показатель промежуточной прецизионности (относительное среднеквадратическое отклонение в условиях вариации факторов «время», «оператор» в одной лаборатории), σRл, % |
Показатель воспроизводимости (относительное среднеквадратическое отклонение воспроизводимости), σR, % |
Показатель точности* (границы относительной погрешности при вероятности Р = 0,95) ±δ, % |
|
Капуста |
|
|
|
|
|
от 0,01 до 0,1 вкл. |
8 |
9 |
11 |
22 |
|
Зерно гороха |
|
|
|
|
|
от 0,01 до 0,1 вкл. |
7 |
8 |
10 |
20 |
|
Зерно сои |
|
|
|
|
|
от 0,01 до 0,1 вкл. |
7 |
8 |
10 |
20 |
|
Масло сои |
|
|
|
|
|
от 0,01 до 0,1 вкл. |
9 |
11 |
13 |
25 |
|
*) соответствует расширенной неопределенности Uотн. при коэффициенте охвата k = 2 |
||||
Полнота извлечения бифентрина, стандартное отклонение, доверительный интервал среднего результата для n = 20, Р = 0,95
|
Анализируемый объект |
Предел обнаружения, мг/кг |
Диапазон определяемых концентраций, мг/кг |
Среднее значение определения, % |
Стандартное отклонение, S, % |
Доверительный интервал среднего результата, ±,% |
|
капуста |
0,01 |
0,01 - 0,1 |
86,3 |
4,9 |
4,4 |
|
Зерно гороха |
0,01 |
0,01 - 0,1 |
89,0 |
3,9 |
3,5 |
|
Зерно сои |
0,01 |
0,01 - 0,1 |
90,5 |
3,8 |
3,4 |
|
Масло сои |
0,01 |
0,01 - 0,1 |
85,8 |
5,2 |
4,7 |
2.3. Избирательность метода
Избирательность метода обеспечивается сочетанием условий подготовки проб и хроматографирования.
3. Средства измерений, реактивы, вспомогательные устройства и материалы
3.1. Средства измерений
|
Хроматограф газовый «Кристалл 2000М» с ДЭЗ и хроматограф газовый Цвет 550 М или аналогичные с ДЭЗ (ДПР) |
|
|
Весы аналитические типа BЛA-200 |
|
|
Весы технические BЛKT-500 |
|
|
Колбы мерные на 10, 100, 1000 см3 |
|
|
Пипетки градуированные |
|
|
Цилиндры мерные на 50 и 100 см3 |
Допускается использование средств измерения с аналогичными или лучшими характеристиками.
3.2. Реактивы
|
Аналитический стандарт бифентрина с содержанием д.в. 97,8 %. |
|
|
Ацетонитрил для ВЭЖХ, «В-230НМ» или хч |
ТУ 6-09-3534-87 |
|
Ацетон, осч |
ТУ 2633-039-44493179-00 |
|
Азот газообразный в баллонах с редуктором |
ТУ-6-16-40-14-88 |
|
Вода дистиллированная |
ГОСТ 6709-79 |
|
Гексан, хч |
ТУ 2631-003-05807999-98 |
|
Дихлорметан, хч |
ТУ 6-09-2662-77 |
|
Натрий сернокислый безводный, ч, свежепрокаленный |
|
|
Натрия хлорид, хч |
|
|
Силикагель для колоночной хроматографии 60 (0,040 - 0,063 мм) (Merck, Германия) |
|
|
Хлороформ, чда |
ТУ-2631-020-11291058-96 |
|
Этилацетат, хч |
ГОСТ 1138-84 |
Допускается использование реактивов квалификацией не ниже указанных.
3.3. Вспомогательные устройства и материалы
|
Колонка капиллярная кварцевая длиной 10 м, внутренним диаметром 0,53 мм с неподвижной фазой НР-1, толщина слоя 1,5 мкм |
|
|
Колонка насадочная стеклянная, длиной 1 м, внутренним диаметром 3 мм |
|
|
Ванна ультразвуковая УЗВ/100 ТН |
|
|
Воронки делительные емкостью 250 и 500 см3 |
|
|
Воронки химические конусные |
|
|
Воронка Бюхнера |
ГОСТ 0147 |
|
Колба Бунзена |
ГОСТ 5614-75 |
|
Колбы-концентраторы емкостью 100 и 250 см3 |
|
|
Колбы плоскодонные емкостью 300 см3 |
|
|
Колонка стеклянная хроматографическая длиной 25 см, диаметром 10 мм |
|
|
Мельница электрическая лабораторная |
ТУ 46-22-236-79 |
|
Микрошприц МШ-10 |
ТУ 2-833-106 |
|
Насос водоструйный |
ГОСТ 10696-75 |
|
Ротационный вакуумный испаритель Buchi R-200/205 (Швейцария) |
|
|
Стаканы химические |
|
|
Стекловата |
|
|
Фильтры бумажные «красная лента» |
ТУ 6.091678-86 |
Допускается использование другого вспомогательного оборудования с аналогичными или лучшими техническими характеристиками.
4. Требования безопасности
4.1. При проведении работы необходимо соблюдать требования техники безопасности, установленные для работ с токсичными, едкими, легковоспламеняющимися веществами (ГОСТ 12.1.005, 12.1.007). Организация обучения работников безопасности труда по ГОСТ 12.0.004.
При выполнении измерений с использованием газового хроматографа и работе с электроустановками соблюдать правила электробезопасности в соответствии с ГОСТ 12.1.019-79 и инструкциями по эксплуатации приборов.
4.2. Помещение лаборатории должно быть оборудовано приточно-вытяжной вентиляцией, соответствовать требованиям пожарной безопасности по ГОСТ 12.1.004-91 и иметь средства пожаротушения по ГОСТ 12.4.009. Содержание вредных веществ в воздухе не должно превышать норм, установленных ГН 2.2.5.1313-03 «Предельно допустимые концентрации (ПДК) вредных веществ в воздухе рабочей зоны».
5. Требования к квалификации операторов
Измерения в соответствии с настоящей методикой может выполнять специалист-химик, имеющий опыт работы методом газожидкостной хроматографии, ознакомленный с руководством по эксплуатации хроматографа, освоивший данную методику и подтвердивший экспериментально соответствие получаемых результатов нормативам контроля погрешности измерений по п. 13.
6. Условия измерений
При выполнении измерений выполняют следующие условия:
• процессы приготовления растворов и подготовки проб к анализу проводят при температуре воздуха (20 ± 5) °С и относительной влажности не более 80 %;
• выполнение измерений на газовом хроматографе проводят в условиях, рекомендованных технической документацией к прибору.
7. Отбор проб и хранение
Отбор проб капусты проводят в соответствии с ГОСТ 1724-85 «Капуста белокочанная свежая. Заготовка и поставка» или ГОСТ 7967-87 «Капуста краснокочанная свежая. Заготовка и поставка».
Отбор проб зерна гороха проводят в соответствии с ГОСТ 28674-90 «Горох. Требования по заготовке и поставке». Для длительного хранения аналитические пробы капусты и зерна гороха помещают в герметично закрытый двойной полиэтиленовый пакет и хранят в морозильной камере с температурой -18 °С.
Отбор проб зерна сои для анализа проводят в соответствии с ГОСТ 10852-86 «Семена масличные. Правила приемки и методы отбора проб».
Пробы зерна просушивают до стандартной влажности и хранят в закрытой стеклянной или полиэтиленовой таре.
Пробы растительного масла хранят в холодильнике при температуре 4 - 6 °С в закрытой стеклянной таре не более 2 месяцев.
8. Подготовка к определению
8.1. Кондиционирование колонки
Капиллярную и или насадочную колонку перед анализом кондиционируют в токе азота при температуре 250 °С до установления нулевой линии.
8.2. Подготовка и очистка реактивов и растворителей
Перед началом работы рекомендуется проверить чистоту применяемых органических растворителей. Для этого 100 см3 растворителя упаривают в ротационном вакуумном испарителе при температуре 40 °С до объема 1,0 см3 и хроматографируют. При обнаружении мешающих определению примесей очистку растворителей производят в соответствии с типовыми методиками.
8.3. Приготовление стандартного и градуировочных растворов
8.3.1. Основной раствор с концентрацией 0,5 мг/см3: точную навеску бифентрина (50 ± 0,5 мг) помещают в мерную колбу вместимостью 100 см3, растворяют в ацетоне и доводят объем до метки гексаном.
Градуировочные растворы бифентрина с концентрациями 0,05, 0,1, 0,25 и 0,5 мкг/см3 готовят методом последовательного разбавления основного раствора 1 по объему, используя гексан.
8.3.2. Раствор № 1 с концентрацией бифентрина 0,5 мкг/см3: в мерную колбу вместимостью 100 см3 вносят 0,1 см3 основного раствора и доводят объем до метки гексаном.
8.3.3. Раствор № 2 с концентрацией бифентрина 0,25 мкг/см3: в мерную колбу вместимостью 10 см3 помещают 5 см3 раствора № 1 и доводят объем до метки гексаном.
8.3.4. Раствор № 3 с концентрацией бифентрина 0,1 мкг/см3: в мерную колбу вместимостью 10 см3 помещают 2 см3 раствора № 1 и доводят объем до метки гексаном.
8.3.5. Раствор № 4 с концентрацией бифентрина 0,05 мкг/см3: в мерную колбу вместимостью 10 см3 помещают 1 см3 раствора № 1 и доводят объем до метки гексаном.
Основной раствор можно хранить в холодильнике при температуре 0 - 4 °С в течение 1 месяца, градуировочные растворы - в течение суток.
Для внесения в образец при определении полноты извлечения используют основной раствор бифентрина, разбавленный гексаном до соответствующей концентрации.
8.4. Построение градуировочного графика
Для построения градуировочного графика в хроматограф вводят по 1 мм3 градуировочных растворов (не менее 3 параллельных измерений для каждой концентрации, не менее 4 точек по диапазону измеряемых концентраций), измеряют высоты или площади пиков и строят график зависимости среднего значения высоты (площади) пика от концентрации бифентрина в градуировочном растворе (мкг/см3).
Градуировочную характеристику необходимо проверять при замене реактивов, хроматографической колонки или элементов хроматографической системы, а также при отрицательном результате контроля градуировочного коэффициента.
Градуировочную зависимость признают стабильной при выполнении следующего условия:
![]()
где С - аттестованное значение массовой концентрации бифентрина в градуировочном растворе;
СК - результат контрольного измерения массовой концентрации бифентрина в градуировочном растворе;
λконтр. - норматив контроля градуировочного коэффициента, % (λконтр. = 10 % при Р = 0,95).
8.5. Подготовка колонки с силикагелем для очистки экстракта
В нижнюю часть стеклянной колонки длиной 25 см и внутренним диаметром 1 см помещают тампон из стекловаты, вносят суспензию 8 г силикагеля в 40 см3 гексана и насыпают 2,5 г безводного сернокислого натрия, дают растворителю стечь до верхнего края сорбента. Колонку промывают 40 см3 гексана со скоростью 1 - 2 капли в секунду, после чего она готова к работе.
8.6. Проверка хроматографического поведения бифентрина на колонке с силикагелем
В подготовленную колонку вносят 1 см3 стандартного раствора бифентрина с концентрацией 0,1 мкг/см3, раствору дают впитаться, после чего через колонку пропускают 20 см3 гексана, элюат отбрасывают. Затем пропускают 80 см3 смеси гексан-этилацетат (95:5), отбирая фракции по 10 см3 каждая, упаривают досуха, остаток растворяют в 1 см3 гексана и анализируют на содержание бифентрина по п. 2.6.8. Фракции, содержащие бифентрин, объединяют, упаривают досуха, остаток растворяют в 1 см3 гексана и анализируют по п. 2.6.8. Рассчитывают содержание бифентрина в элюате, определяя полноту вымывания вещества из колонки и необходимый для этого объем элюента.
Примечание: профиль вымывания бифентрина может меняться при использовании новой партии сорбента.
8.7. Подготовка приборов и средств измерения
Установка и подготовка всех приборов и средств измерения проводится в соответствии с требованиями технической документации.
9. Проведение определения
9.1. Экстракция бифентрина из капусты
Анализируемые объекты измельчают, берут навеску (10 ± 0,1) г, помещают в коническую колбу на 250 см3, проводят экстракцию 50 см3 60 %-го водного ацетона на ультразвуковой бане в течение 15 мин, экстракцию повторяют дважды порциями по 40 см3. Объединенный экстракт фильтруют через бумажный фильтр «красная лента», фильтр обмывают 10 см3 водного ацетона, половину объединенного экстракта отбрасывают. Оставшуюся часть экстракта (эквивалентную 5 г образца) упаривают на роторном испарителе при температуре не выше 40 °С до водного остатка, который переносят в делительную воронку на 250 см3 и добавляют равный объем 10 %-го раствора хлорида натрия, после чего проводят экстракцию бифентрина хлороформом трижды порциями по 30 см3. После полного расслоения жидкостей* водную фазу отбрасывают, хлороформные экстракты фильтруют через слой безводного сульфата натрия (10 г) в колбу-концентратор объемом 250 см3, осушитель промывают 10 - 15 см3 хлороформа. Экстракт упаривают на роторном испарителе при температуре 50 - 60 °С. Сухой остаток растворяют в 5 см3 гексана и 1 мм3 вводят в испаритель хроматографа. При наличии на хроматограмме пиков коэкстрактивных веществ, мешающих определению бифентрина, проводят очистку по п. 9.5.
________________
* При образовании сравнительно стойких эмульсий для ускорения расслаивания добавляют 10 - 15 см3 насыщенного раствора хлорида натрия и/или 2 - 5 см3 метанола.
9.2. Экстракция бифентрина из зерна гороха
Навеску зерна гороха массой (10 ± 0,1) г, размолотого на лабораторной мельнице, помещают в коническую колбу на 100 см3 и экстрагируют бифентрин 40 см3 ацетона на ультразвуковой бане в течение 15 мин, гомогенат фильтруют через фильтр «красная лента» в мерный цилиндр на 250 см3, экстракцию повторяют дважды по 30 см3 ацетона. Фильтр обмывают 10 см3 ацетона. Объединенные экстракты перемешивают и количественно переносят в коническую колбу, которую помещают в морозильную камеру с температурой -18 °С на 2 - 3 ч. После вымораживания экстракт фильтруют через бумажный фильтр «красная лента», фильтр и колбу обмывают по 10 см3 охлажденного ацетона. К содержимому колбы добавляют 40 см3 воды так, чтобы соотношение растворителей ацетон-вода составляло 3:2. Экстракцию пестицида хлороформом проводят трижды порциями по 40 см3 в делительной воронке на 500 см3. После полного разделения фаз хлороформный экстракт фильтруют через слой безводного сульфата натрия (10 г) в колбу-концентратор на 250 см3, осушитель промывают 10 - 15 см3 хлороформа. Полученный экстракт выпаривают на роторном испарителе при температуре 50 - 60 °С. Сухой остаток растворяют в 2 см3 гексана на ультразвуковой бане и хроматографируют. При необходимости проводят очистку по п. 9.5.
9.3. Экстракция бифентрина из зерна сои
Берут навеску зерна сои (10 ± 0,1) г, измельчают его в фарфоровой ступке и количественно переносят в коническую колбу на 300 см3, добавляют 100 см3 80 %-го водного ацетонитрила и экстрагируют бифентрин на ультразвуковой бане в течение 5 мин. Экстракцию повторяют дважды порциями по 50 см3. Объединенный экстракт фильтруют через бумажный фильтр «красная лента» в чистую колбу на 250 см3, фильтр обмывают дважды 20 см3 водного ацетонитрила. Фильтрат переносят в делительную воронку на 500 см3, добавляют 30 см3 гексана и встряхивают воронку в течение 2 - 3 мин. После разделения фаз гексан отбрасывают, ацетонитрильный слой возвращают в делительную воронку и повторяют промывку гексаном дважды порциями по 30 см3. Водно-ацетонитрильный экстракт переносят в колбу-концентратор объемом 250 см3 и упаривают на роторном испарителе при температуре 50 - 60 °С до водного остатка. Водный остаток переносят в делительную воронку объемом 250 см3, колбу обмывают 30 см3 10 %-го хлористого натрия и смыв добавляют к водному остатку. Затем в делительную воронку добавляют 50 см3 дихлорметана, встряхивают 2 - 3 мин и оставляют до полного разделения фаз. Органическую фазу фильтруют в колбу-концентратор через фильтр «красная лента» со слоем безводного сернокислого натрия. Экстракцию бифентрина из водной фазы повторяют дважды порциями дихлорметана по 30 см3. Объединенный экстракт упаривают на роторном испарителе при температуре не выше 40 °С до сухого остатка. Сухой остаток растворяют в 1 см3 гексана и 1 мм3 вводят в испаритель хроматографа. При наличии на хроматограмме пиков коэкстрактивных веществ, мешающих определению бифентрина, проводят очистку по п. 9.5.
9.4. Экстракция бифентрина из масла сои
Навеску масла (10 ± 0,1) г растворяют в 100 см3 гексана в плоскодонной колбе объемом 250 см3 в ультразвуковой ванне в течение 5 мин и оставляют на час. Затем экстракт количественно переносят в делительную воронку объемом 250 см3, добавляют 100 см3 ацетонитрила и встряхивают воронку в течение 2 - 3 мин. После разделения фаз ацетонитрильный слой собирают, пропуская через фильтр «красная лента» со слоем безводного сульфата натрия в мерный цилиндр на 250 см3. Экстракцию ацетонитрилом повторяют дважды порциями по 50 см3. Объединенный экстракт переносят в чистую делительную воронку объемом 250 см3 и ацетонитрильный слой трижды промывают гексаном порциями по 15 - 20 см3. Гексановый слой отбрасывают, а промытый ацетонитрильный экстракт упаривают до сухого остатка на роторном испарителе при температуре не выше 50 - 60 °С.
Сухой остаток растворяют в 1 см3 гексана и хроматографируют. При наличии мешающих примесей дополнительную очистку проводят на колонке с силикагелем по п. 9.5.
9.5. Очистка экстрактов на колонке с силикагелем
Количественно переносят в кондиционированную хроматографическую колонку (п. 8.5) 1 см3 полученного по пп. 9.1 - 9.4 экстракта, дают впитаться, после чего через колонку пропускают 20 см3 гексана, элюат отбрасывают. Затем колонку промывают 30 см3 смеси гексан-этилацетат (95:5). Первые 20 см3 элюата отбрасывают, следующие 10 см3 собирают в колбу-концентратор на 50 см3, выпаривают досуха на роторном испарителе, растворяют в 1 см3 гексана и хроматографируют.
9.6. Условия хроматографирования
9.6.1. Хроматографирование экстрактов капусты и зерна гороха
Хроматограф Цвет-550М с ДПР. Колонка насадочная стеклянная, длиной 1 м, внутренним диаметром 3 мм. Температура колонки 205 °С, испарителя 230 °С, детектора 300 °С. Расход газа-носителя (азот) через колонку 43,5 см3/мин. Навеска 10 г. Конечный объем экстракта 2 см3. Дозируемый объем 1 мм3. Время удерживания бифентрина (3,35 ± 0,01) мин.
9.6.2. Хроматографирование экстрактов зерна и масла сои
Хроматограф газовый «Кристалл 2000М» с ДЭЗ.
Колонка капиллярная кварцевая длиной 10 м, внутренним диаметром 0,53 мм с неподвижной фазой НР-1, толщина слоя 1,5 мкм.
Температура колонки 200 °С, температура испарителя 230 °С, детектора 300 °С.
Расход газа-носителя (азот) через колонку Г1 - 8,4 см3/мин (давление на входе 105 кПа), Г2 - 41,5 см3/мин, Г3 - 20 см3/мин. Деление потока 1:5. Объем вводимой пробы 1 мм3.
Время удерживания бифентрина (4,97 ± 0,08) мин.
10. Обработка результатов анализа
Количественное определение проводят методом абсолютной калибровки, содержание бифентрина в пробе (X, мг/кг) вычисляют по формуле:
![]()
где H1 - высота (площадь) пика бифентрина в стандартном растворе, мм (мв∙с);
Н2 - высота (площадь) пика бифентрина в анализируемой пробе, мм (мв∙с);
V - объём экстракта, подготовленного для хроматографирования, см3;
Р - навеска анализируемого образца, г;
С - концентрация бифентрина в стандартном растворе, мкг/см3.
Содержание остаточных количеств бифентрина в анализируемом образце вычисляют как среднее из 2-х параллельных определений.
Образцы, дающие пики большие, чем стандартный раствор бифентрина 1 мкг/см3, разбавляют.
11. Проверка приемлемости результатов параллельных определений
За результат анализа принимают среднее арифметическое результатов двух параллельных определений, расхождение между которыми не превышает предела повторяемости (1):
|
|
(1) |
где X1 и Х2 - результаты параллельных определений, мг/кг;
r - значение предела повторяемости (r = 2,8σr).
При невыполнении условия (1) выясняют причины превышения предела повторяемости, устраняют их и вновь выполняют анализ.
12. Оформление результатов
Результат анализа представляют в виде:
(![]() ± Δ)
мг/кг при вероятности Р = 0,95, где
± Δ)
мг/кг при вероятности Р = 0,95, где
![]() -
среднее арифметическое результатов определений, признанных приемлемыми, мг/кг;
-
среднее арифметическое результатов определений, признанных приемлемыми, мг/кг;
Δ - граница абсолютной погрешности, мг/кг.
![]()
где δ - граница относительной погрешности методики (показатель точности в соответствии с диапазоном концентраций), %.
В случае если содержание компонента менее нижней границы диапазона определяемых концентраций, результат анализа представляют в виде:
содержание вещества в пробе «менее нижней границы определения» (например: менее 0,01*,
где * - 0,01 мг/кг - предел определения бифентрина в капусте, зерне гороха, сои и масле сои).
13. Контроль качества результатов измерений
Оперативный контроль погрешности и воспроизводимости измерений осуществляется в соответствии с ГОСТ Р ИСО 5725-1-6-2002 «Точность (правильность и прецизионность) методов и результатов измерений».
13.1. Стабильность результатов измерений контролируют перед проведением измерений, анализируя один из градуировочных растворов.
13.2. Плановый внутрилабораторный оперативный контроль процедуры выполнения анализа проводится с применением метода добавок.
Величина добавки Cд должна удовлетворять условию:
Cд = Δл,x + Δл,x' где
±Δл,x (±Δл,x') - характеристика погрешности (абсолютная погрешность) результатов анализа, соответствующая содержанию компонента в испытуемом образце (расчетному значению содержания компонента в образце с добавкой соответственно), мг/кг; при этом:
Δл = ±0,84∙Δ, где
Δ - граница абсолютной погрешности, мг/кг:
![]()
δ - граница относительной погрешности методики (показатель точности в соответствии с диапазоном концентраций), %.
Результат контроля процедуры Kк рассчитывают по формуле:
Kк = Х' - Х - Сд, где
X', X, Сд - среднее арифметическое результатов параллельных определений (признанных приемлемыми по п. 11) содержания компонента в образце с добавкой, испытуемом образце, концентрация добавки, соответственно, мг/кг (мг/дм3).
Норматив контроля K рассчитывают по формуле:
Проводят сопоставление результата контроля процедуры (Kк) с нормативом контроля (K).
Если результат контроля процедуры удовлетворяет условию
|
|Kк| ≤ K |
(2) |
процедуру анализа признают удовлетворительной.
При невыполнении условия (2) процедуру контроля повторяют. При повторном невыполнении условия (2) выясняют причины, приводящие к неудовлетворительным результатам, и принимают меры к их устранению.
13.3. Проверка приемлемости результатов измерений, полученных в условиях воспроизводимости:
Расхождение между результатами измерений, выполненных в двух разных лабораториях, не должно превышать предела воспроизводимости (R):
|
|
(3) |
где X1 и Х2 - результаты измерений в двух разных лабораториях, мг/кг;
R - предел воспроизводимости (в соответствии с диапазоном концентраций), %.
14. Разработчики
Долженко В.И., Цибульская И.А., Карпова Л.М. (ГНУ Всероссийский научно-исследовательский институт защиты растений, Санкт-Петербург).
Методика прошла метрологическую экспертизу (Свидетельство об аттестации № 01.5.04.008) и внесена в Федеральный реестр (ФР.1.31.2011.10398).