Государственный
комитет санитарно-эпидемиологического
надзора Российской Федерации
Федеральные санитарные правила, нормы, гигиенические нормативы
ОПРЕДЕЛЕНИЕ ЛЕТУЧИХ N-НИТРОЗАМИНОВ
В ПРОДОВОЛЬСТВЕННОМ СЫРЬЕ И ПИЩЕВЫХ ПРОДУКТАХ
Методические указания по методам контроля
МУК 4.4.1.011-93
Москва
1993
Федеральные санитарные правила, нормы и гигиенические нормативы:
нормативные акты, устанавливающие критерии безопасности и (или) безвредности для человека факторов среды его обитания и требования к обеспечению благоприятных условий его жизнедеятельности;
обязательны для соблюдения всеми государственными органами и общественными объединениями, предприятиями или иными хозяйствующими субъектами, организациями и учреждениями, независимо от их подчиненности и форм собственности, должностными лицами и гражданами;
должностные лица и граждане Российской Федерации, допустившие санитарное правонарушение, могут быть привлечены к дисциплинарной, административной и уголовной ответственности. Закон Российской Федерации «О санитарно-эпидемиологическом благополучии населения» от 19 апреля 1991 г. Ведомости съезда народных депутатов и Верховного Совета Российской Федерации, 1991. № 20. С. 641.
|
«УТВЕРЖДАЮ» Председатель Государственного комитета санитарно-эпидемиологического надзора Российской федерации, Главный государственный врач Российской Федерации _________________________ Е.Н. Беляев «22»___декабря___ 1993 года |
Дата введения с момента утверждения
Определение летучих N-нитрозаминов в продовольственном сырье и пищевых продуктах.
Методические указания по методам контроля.
ОБЩИЕ ПОЛОЖЕНИЯ
Настоящие методические указания распространяются на продовольственное сырье и пищевые продукты и устанавливают флуориметрический хемилюминисцентный методы определения летучих N-нитрозаминов.
Методические указания предназначены для учреждений санэпиднадзора и других ведомств, осуществляющих контроль качества и безопасность продовольственного сырья и пищевых продуктов.
ФЛЮОРИМЕТРИЧЕСКИЙ МЕТОД
1. СУЩНОСТЬ МЕТОДА
В основу метода положено Авторское свидетельство «Способ определения N-нитрозаминов в пищевых продуктах» № 1157449-1985 Б.И. № 1 1985 г.
Метод определения N-нитрозаминов в пищевых продуктах и продовольственном сырье состоит в выделении летучих N-нитрозаминов (НА) путем перегонки с паром или в вакууме; экстракции хлористым метиленом НА из водного дистиллята; концентрации экстракта; денитрозировании НА бромистым водородом в уксусной кислоте; алкилировании образовавшихся аминов 8-метокси-5-хинолинсульфонилазиридином (КАЗ), разделении и количественном определении образовавшихся флуоресцирующих 8-метокси-5-[N-(2-N-диэтиламино)]хинолинсульфонамидных производных (КАЭ-производные) в тонком слое силикагеля.
Идентификацию НА осуществляют сравнением подвижности в тонком слое силикагеля флуоресцирующих КАЭ-производных из образца с подвижностью соответствующих стандартных производных: диметиламина - КАЭ-ДМА, диэтиламина - КАЭ-ДЭА, дипропиламина - КАЭ-ДПА.
В основе полуколичественного определения лежит визуальное сравнение интенсивности флуоресценции пятен КАЭ-производных из образца с интенсивностью флуоресценции пятен стандартных соединений. При необходимости возможно также количественное определение путем измерения величины флуоресценции КАЭ-производных.
Нижний предел определения НА - 1 мкг/кг продукта.
2. ОТБОР ПРОБ
Навеска массой 100 - 300 г (или см3) отбирается из средней пробы, подготовленной в соответствии с действующими НТД на конкретный вид продукции. Отобранные образцы можно хранить в морозильной камере при температуре -8 - -18 °С от 1 до 3-х суток в зависимости от сроков реализации продукции.
3. АППАРАТУРА, МАТЕРИАЛЫ, РЕАКТИВЫ
Весы технические по ГОСТ 19491-74 и весы аналитические по ГОСТ 24104-80.
Шкаф сушильный, нагревательные приборы (электронагреватель по ГОСТ 13268-83, колбонагреватель или электроплитка по ГОСТ 14919-83).
Ротационный испаритель с ловушкой по ТУ 25-11-917-76.
Насос водоструйный по ГОСТ 10696-75.
Контактные термометры по ГОСТ 9871-75.
Прибор для флуоресцентного анализа витаминов в растворе (модель 833) МРТУ 64-1-1080-63 или диагностическая лампа ОЛД-41.
Флуориметр ЭФ-3М с фильтрами B1-1, B2-1 или другие спектрофлуориметры с аналогичными или более высокими оптическими характеристиками.
Камера для ТСХ с притертыми крышками, например, стеклянные четырехугольные сосуды 195´195´200 мм завода «Дружная горка».
Пластинки для ТСХ «Силуфол» (Чехо-Словакия) или «Сорбфил», ПКБ «Пластмаш» (без флуоресцентного индикатора).
Микрошприц МШ-10 на 10 мкл или калиброванные стеклянные капилляры.
Мясорубка или гомогенизатор по ГОСТ 15906-79.
Штативы Бунзена.
Бани глицериновые, водяные.
Дефлегматор елочный по ГОСТ 20729-75
Цилиндры мерные на 50, 100, 200 мл по ГОСТ 1770-74.
Пипетки емкостью 1, 2 мл по ГОСТ 20292-74.
Колбы мерные или пробирки мерные 2, 5, 25, 50 мл по ГОСТ 1770-74.
Холодильник Либиха с алонжем по ГОСТ 9499-70.
Холодильник обратный по ГОСТ 23932-79 и 25336-82.
Насадка для паровика изготовлена по эскизу, см. рис. 1. (поз. 8).
Колбы плоскодонные на 250 мл по ГОСТ 10394-72.
Колбы круглодонные на 1000 500 и 5 мл по ТУ 48-52.
Делительные воронки емкостью 5 и 250 мл по ГОСТ 23932-79 и ГОСТ 25336-82.
Эксикатор.
Аммиак водный по ГОСТ 3760-79.
Ацетонитрил по ТУ 6-09-3534-74.
Бензол по ГОСТ 5955-75.
Вода дистиллированная по ГОСТ 6709-72
Гексан-н по ТУ 6-09-5362-68
Кальций хлористый прокаленный
Кислота бромистоводородная по ГОСТ 2062-76
Кислота соляная по ГОСТ 3118-77 и водный раствор 0,1 Н
Кислота серная по ГОСТ 4204-77 и водный раствор 1 Н, 5 Н
Кислота уксусная ледяная по ГОСТ 61-75
Метилен хлористый, МРТУ 6-09-5362-68
Масло вазелиновое, парафиновое
Натрия гидроокись по ГОСТ 4328-77, хч и водный раствор 0,1 Н
Натрий сернокислый прокаленный по ГОСТ 18344-78, хч
Натрий углекислый кислый по ГОСТ 4201-79
Натрий хлористый по ГОСТ 4233-77
Спирт метиловый по ГОСТ 6995-77
Спирт этиловый по ГОСТ 18300-87
Сульфаминовая или сульфаниловая кислота по ГОСТ 5282-78;
Стандартные и рабочие растворы:
8-метокси-5-хинолинсульфонилазиридин (КАЗ)*
8-метокси-5-[N-(2-N-диэтиламино)]хинолинсульфонамидые производные аминов: диметиламина (КАЭ-ДМА), диэтиламина (КАЭ-ДЭА), дипропиламина (КАЭ-ДПА) - спиртовые растворы концентрации C1 - 2 мг/мл.
2 - 3 % раствор газообразного бромистого водорода в ледяной уксусной кислоте (HBr/АсОН)
Нитрозодипропиламин (НДПА) - гексановый раствор, С = 0,2 мг/100 мл
_________
Примечание: КАЗ (в концентрации 1 мг/5 мл), КАЭ-ДМА, КАЭ-ДЭА, КАЭ-ДПА (в концентрации 5 мг/5 мл) можно приобрести в Институте биологической и медицинской химии РАМН по адресу: Москва, 119832, ул. Погодинская, д. 10. Обращаться по тел. 246-58-20
- Грачева Ирина Николаевна
- Точилкин Анатолий Иванович
- Ковельман Инна Рафаиловна.
4. ТРЕБОВАНИЯ ТЕХНИКИ БЕЗОПАСНОСТИ ПРИ ПРОВЕДЕНИИ ИСПЫТАНИЙ.
Помещение, в котором производится определение нитрозаминов обязательно должно быть оборудовано приточно-вытяжной вентиляцией.
Работу с нитрозаминами, стандартами, другими химическими соединениями и растворителями проводить в вытяжном шкафу с использованием индивидуальных средств защиты (очки, перчатки и др.).
В лаборатории, где проводятся работа с канцерогенными летучими НА необходимо всегда иметь 2 - 3 % раствор газообразного HBr в уксусной кислоте для разрушения НА при попадании их на рабочие места и пол. В целях разрушения летучих НА в воздухе по окончании работы помещение необходимо обработать ультрафиолетовым светом.
Транспортировка и хранение НА осуществляется в стеклянных запаянных ампулах, обернутых в асбестовую ткань и упакованных в металлическую тару, которую также заплавляют парафином.
НА хранят в специальном холодильнике в отсутствии анализируемых проб.
5. ПОДГОТОВКА К ИСПЫТАНИЮ.
5.1. Приготовление растворов
5.1.2. Стандартный раствор НДПА с концентрацией 2 мкг/мл, используется при анализе в качестве внутреннего стандарта.
5.1.3. Стандартные растворы КАЭ-ДМА, КАЭ-ДЭА к КАЭ-ДПА. 2,5 мл содержимого ампул растворов КАЭ-ДМА, КАЭ-ДЭА и КАЭ-ДПА (концентрации C1 = 5 мг/5 мл)* переносят в мерные колбы на 25 мл, доводят до метки этиловым спиртом и получают растворы с концентрацией С2 = 0,1 мг/мл. Для получения смеси стандартов в мерную колбу на 25 мл переносят оставшуюся часть содержимого ампул (2,5 мл каждого КАЭ-производного)*, раствор доводят до метки этиловым спиртом. Концентрация раствора каждого стандартного производного в этой смеси равна С2 = 0,1 мг/мл. Для получения смеси стандартов меньших концентрация С3 и С4 в мерные колбы на 25 мл помещают соответственно 2,5 и 1,25 мл стандартного раствора С2 и растворы доводят до метки этиловым спиртом. Концентрация каждого стандарта в этих смесях соответственно равна С3 = 10 нг/мкл и С4 = 5 нг/мкл. Рабочий раствор КАЗ (п. 5.1.1.) и стандартные растворы КАЭ-производных (п. 5.1.3.) хранить в темной посуде с пришлифованной пробкой в холодильнике не более 1 года.
5.1.6. Подготовка к анализу растворителей и реактивов, контроль их чистоты.
5.1.6.1. Подготовка к работе хлористого метилена. Предназначенный для использования хлористый метилен предварительно подвергается фракционной перегонке. 60 - 75 мл хлористого метилена (количество, используемое при анализе) концентрируют до 1 мл по п. 6.1. и анализируют концентрат на содержание НА флюориметрическим или хемилюминисцентным методом в соответствии с п. 7 или п. 12. При отсутствии НА хлористый метилен может быть использован в работе; при обнаружении НА хлористый метилен подвергают дополнительной очистке. Для этого 500 мл хлористого метилена дважды промывают концентрированной серной кислотой (2 раза по 30 мл) и дистиллированной водой, затем 30 % раствором гидроокиси натрия и вновь дистиллированной водой до нейтрального рН, высушивают прокаленным хлористым кальцием и проводят фракционную перегонку, собирая фракцию при температуре 40° ± 0,2 °С.
5.1.6.2. Контроль чистоты реактивов. Пробу реактивов (Na2SO4, NaCl, NaOH и др.) каждой партии в количестве, используемом в эксперименте, выдерживают раздельно в течение 10 мин с 60 - 70 мл хлористого метилена, отфильтровывают, концентрируют по п. 6.1. до 1 мл и анализируют концентрат на наличие НА. При отсутствии НА - реагенты используются в работе, при наличии НА - подвергаются дополнительной очистке путем перекристаллизации или заменяются на новую партию.
6. ВЫДЕЛЕНИЕ НИТРОЗАМИНОВ.
6.1. Выделение нитрозаминов путем перегонки с паром. Навеску 50 - 100 г измельченного в мясорубке или гомогенизаторе пищевого продукта помещают в круглодонную колбу на 500 мл, соединенную с паровиком и прямым холодильником (рис. 1). К продукту добавляют 10 г хлористого натрия, 10 г сульфата натрия или магния, 25 - 50 мл дистиллированной воды (в зависимости от влажности продукта), 5 мл 2 % раствора сульфаминовой (или сульфаниловой) кислоты (п. 5.1.5.), 5 - 10 мл 1Н раствора серной кислоты до рН < 3,0, 1 мл гексанового раствора ДПНА (п. 5.1.2.) и НА отгоняют с водяным паром, собирая 200 - 230 мл дистиллята. Если перегонка с паром сопровождается вспениванием массы (пиво, зерно, гидробионты и продукты вырабатываемые из них), добавляют 5 - 10 мл насыщенного раствора апиезона в изопропиловом спирте, а в случае спекания продуктов в процессе дистилляции (хлеб, зерно и др.) - мелкие керамические крошки или кусочки стеклянных трубочек. Для наиболее полного выделения НА из алкогольных напитков необходимо разбавить образец водой до получения 20 % концентрации в нем спирта. Дистиллят переносят в делительную воронку и нитрозамины экстрагируют свежеперегнанным хлористым метиленом 4 - 5 раз по 15 мл.
ПРИМЕЧАНИЕ: использование других неполярных растворителей: эфир, этилацетат, гексан, хлороформ и др. не рекомендуются, т.к. степень извлечения нитрозаминов из водных растворов очень низка.
Объединенные хлорметиленовые экстракты высушивают прокаленным сульфатом магния, отфильтровывают, осушитель промывают небольшим объемом хлористого метилена, объединенные фильтраты помещают в круглодонную колбу на 100 - 150 мл, снабжают дефлегматором (или воздушным холодильником), помещают в водяную баню и упаривают хлористый метилен до 5 - 7 мл выдерживанием при температуре бани 55 - 65 °С. Целесообразно пропускание тока инертного газа через хлорметиленовый раствор. По достижении объема раствора 5 - 7 мл дефлегматор смывают 2 мл гексана и продолжают концентрировать пробу до 1 - 2 мл.
6.2. Выделение нитрозаминов перегонкой в вакууме. Пробу исследуемого продукта массой 20 - 50 г, отвешенную с погрешностью 0,1 г, помещают в круглодонную колбу вместимостью 200 мл, добавляют 75 - 80 мл дистиллированной воды, 1 мл гексанового раствора N-нитрозодипропиламина (НДПА), 4 мл 0,1 Н раствора гидроокиси натрия и 20 мл парафинового или вазелинового масла для предотвращения перебрасывания массы во время дистилляции и присоединяют к вакуумной системе (рис. 2).
Колбу с исследуемым продуктом помещают на песочную баню, под которой устанавливается газовая горелка; приемную колбу охлаждают смесью сухого льда и гексана (ацетона) или жидким азотом. Установив в системе вакуум до 1 атм. проводят дистилляцию около 1 часа при температуре бани 98 - 100 °С до получения 50 мл дистиллята. Чтобы избежать потери НА желательно проводить дистилляцию в закрытой системе, как можно реже подключая ее к вакуумному насосу. Герметичность системы можно сохранить используя чистую вакуумную смазку или тефлоновые манжеты для шлифов. После окончания дистилляции снимают обогрев, охлаждение, отсоединяют шланг вакуумной системы, наконечник приемника закрывают тефлоном или пробкой. После оттаивания приемной колбы систему перегонки разбирают, насадку ополаскивают несколькими миллилитрами дистиллированной воды, которые добавляют к дистилляту. В круглодонную колбу с дистиллятом добавляют 1 - 2 мл этанола, 0,5 мл 5 Н серной кислоты и экстрагируют трижды по 7 - 10 мл хлористым метиленом. Последний собирают пипеткой со дна колбы и переносят в стаканчик емкостью 50 мл с 1 - 2 г безводного сульфата натрия. После подсушивания каждую порцию экстракта переносят в пробирку. Оставшийся в стаканчике сульфат натрия промывают 2 мл хлористого метилена, который также переносится в пробирку с экстрактом. Концентрирование экстракта проводят на водяной бане при температуре 40 °С под током азота до 5 - 7 мл, затем экстракт переносят в центрифужную пробирку и концентрируют под током азота при комнатной температуре до 1 мл (не допускать полной отгонки экстракта!).
7. ПРОВЕДЕНИЕ АНАЛИЗА.
7.1. Проведение алкилирования.
7.1.1. Проведение денитрозирования. Полученный по п. 6.1. или п. 6.2. гексановый или хлорметиленовый раствор (2 мл) делят на две равные части (раствор А и раствор Б). К 1 мл раствора (раствор А) добавляют 1 мл 2 - 3 % раствора газообразного HBr в уксусной кислоте (HBr/АсОН) (НЕЛЬЗЯ ИСПОЛЬЗОВАТЬ БРОМИСТОВОДОРОДНУЮ КИСЛОТУ!) и выдерживают этот раствор в течение 30 мин при комнатной температуре. Далее раствор упаривают досуха на роторном испарителе при 40 - 50 °С и Р = 10 - 15 мм рт.ст.
7.1.2. Получение КАЭ-производных. К остатку после упаривания на роторном испарителе добавляют 0,2 - 0,3 мл раствора кислого углекислого натрия (п. 5.1.4.) до рН > 8, 0,5 мл спиртового раствора КАЭ (п. 5.1.1.), кипятят 30 мин с обратным холодильником на глицериновой бане при температуре 100 °С. При определении содержания нитрозаминов в солоде и пиве добавляют 2,5 мл рабочего раствора КАЭ (п. 5.1.1.). Смесь упаривают досуха, остаток растворяют в 0,3 - 0,5 мл этилового спирта. Этот раствор используют для анализа нитрозаминов. Полученные КАЭ-производные аминов, содержание которых эквивалентно количеству нитрозаминов в пробе, анализируют методом ТСХ в сравнении со стандартами, приготовленными по п. 5.1.3. Для проведения «холостой пробы» ко второй части хлорметиленового экстракта (раствор Б) добавляют 0,1 мл рабочего раствора КАЭ (п. 5.1.1.), 0,2 мл раствора кислого углекислого натрия (п. 5.1.4.), кипятят 30 мин. Обрабатывают аналогично раствору А. Полученный спиртовой раствор («холостая проба») анализируют методом ТСХ, результаты которой учитывают при расчете содержания нитрозаминов.
7.1.3. Тонкослойная хроматография.
7.1.3.1. Подготовка пластинок для хроматографического анализа.
Пластинку помещают в камеру для хроматографии, в которую предварительно наливают смесь бензол-этилацетат-уксусная кислота (100:50:1) (система 1). Развитие хроматограмм проводят до достижения растворителем верхнего края пластинки. Затем пластинку извлекают из камеры, сушат 10 мин на воздухе, помещают в сушильный шкаф и активируют при 110 - 120 °С в течение часа. Для анализа требуется несколько пластинок, которые после активирования до использования хранят в закрытом эксикаторе над прокаленным хлористым кальцием.
7.1.3.2. Проведение тонкослойной хроматографии, идентификация нитрозаминов и определение.
На расстоянии 3 см от нижнего края пластинки проводят карандашом тонкую горизонтальную линию, на которую с интервалом в 1 - 3 см наносят 10 точек. В первую точку наносят 20 - 30 мкл КАЭ-производных исследуемого раствора А (п. 7.1.2.), во вторую - 20 - 30 мкл «холостой пробы». В третью и четвертую точки наносят соответственно по 1 мкл растворов КАЭ-ДМА и КАЭ-КЭА в концентрации С2; в пятую, шестую и седьмую точки - соответственно по 1 мкл растворов КАЭ-ДМА, КАЭ-ДЭА и КАЭ-Д в концентрации С3; в восьмую и девятую точки - соответственно растворы КАЭ-ДМА и КАЭ-ДЭА в концентрации С4. Пластинку поворачивают на 180° и помещают в хроматографическую камеру с системой 1. Развитие хроматограммы проводят до достижения фронтом верхнего края пластинки. Пластинку вынимают из камеры, сушат под тягой на воздухе 15 минут срезают 2 см ниже фронта, тем самым удаляя липофильные примеси. Пластинку вновь переворачивают на 180° и помещают в хроматографическую камеру со смесью ацетонитрил - 25 % аммиак (9:1) (система 2). Уровень растворителя должен быть примерно на 1 см ниже нанесенных пятен. Развитие хроматограммы проводят до достижения растворителем верхнего края пластинки. Пластинку сушат на воздухе и вновь помещают в хроматографическую камеру с системой 1. Хроматографируют в том же направлении, сушат и рассматривают в УФ-свете (лампа ОЛД-41 или прибор для флуоресцентного анализа витаминов, модель 833). Сравнивая подвижность флуоресцирующих производных, которые обнаружены в образце, с подвижностью соответствующих КАЭ-производных стандартов, осуществляют идентификацию нитрозаминов.
Значения Rf КАЭ-производных
|
Соединения |
Rf |
|
КАЭ-ДЭА |
0,21 |
|
КАЭ-ДМА |
0,25 |
|
КАЭ-ДПА |
0,62 |
Сравнивая интенсивность флуоресценции различных количеств стандартных нитрозаминов с интенсивностью флуоресценции образца и «холостой пробы» определяют количество нитрозаминов в пятне. Содержание нитрозаминов в пробе продукта рассчитывают по формуле. При этом учитывается степень извлечения нитрозаминов из образца (%) путем сравнения интенсивности флуоресценции внутреннего стандарта (НДПА), вносимого в продукт (по п. 6.1.) с интенсивностью стандарта - НДПА (Rf = 0,62).
![]()
где К - количество нитрозаминов в продовольственном сырье и пищевом продукте, мкг/кг,
N - количество нитрозаминов в пятне с учетом «холостого опыта», мкг,
П - навеска продукта, кг,
V1 - объем анализируемой пробы, мкл,
V2 - объем нанесенной пробы, мкл,
2,4 - поправочный коэффициент,
% - степень извлечения нитрозаминов из образца.
При необходимости количественное определение содержания нитрозаминов в продовольственном сырье и пищевых продуктах проводят на флуориметре. На двух или более пластинках (в зависимости от вязкости анализируемого раствора) на расстояния 3 см от нижнего края пластинки наносят полосой в 6 - 7 см весь (100 мкл) анализируемый раствор и на тех же пластинках полосой в 3 - 4 см наносят по 100 мкл растворов стандартов с концентрацией, которая обнаружена в пробе. Аналогично наносят пробы «холостого опыта». Хроматографируют как описано выше, пункт 7.1.3.2. Пластинки высушивают на воздухе, рассматривают в УФ свете, отмечают иглой или карандашом флуоресцирующие зоны исследуемого раствора и стандартов, объединяют их вместе с каждой пластинки, разрезают на мелкие кусочки, помещают в колбочки на 10 мл, заливают 4 мл 1Н раствора НСl и, периодически встряхивая, выдерживают 30 минут при комнатной температуре. Раствор осторожно сливают в чистые колбочки (если попадают частицы силикагеля, пробы фильтруют), ополаскивают силикагель 3 раза по 1 мл 1Н НСl и промывные воды объединяют с основным раствором. Аналогично поступают с каждой флуоресцирующей зоной образца, стандартов и «холостого опыта».
Величину флуоресценции полученных растворов (по 7 мл каждого) измеряют на спектрофлуориметре при длине волны возбуждения 340 нм и длине волны флуоресценции 480 нм.
Количество нитрозаминов в образце рассчитывают по формуле:
![]()
где К - количество соответствующего нитрозамина в продовольственном сырье и пищевом продукте, мкг/кг,
П - количество нитрозаминов стандарта, мкг,
М - навеска продукта, кг,
Ф1 - флуоресценция нитрозаминов образца за вычетом флуоресценции «холостого опыта»,
Ф2 - флуоресценция соответствующего нитрозаминового стандарта,
2,4 - поправочный коэффициент,
% - степень извлечения нитрозаминов из образца.
Статистическая обработка результатов анализа продовольственного сырья и пищевых продуктов показала, что относительное стандартное отклонение колеблется от 20 до 30 % (при n = 2, P = 0,90).
ХЕМИЛЮМИНИСЦЕНТНЫЙ МЕТОД
(Арбитражный метод анализа)
8. СУЩНОСТЬ МЕТОДА
Метод идентификации и количественного определения НА состоит в выделении летучих НА путем перегонки с паром или в вакууме, экстракции хлористым метиленом НА из водного дистиллята, концентрировании экстракта, разделении смеси методом газо-жидкостной хроматографии и количественном определении модифицированных НА с помощью высокоселективного и высокочувствительного хемилюминисцентного (термоэнергетического) детектора ТЕА-502.
Идентификацию НА осуществляют по временам удерживания в сравнении с параметрами удерживания стандартных НА. Количественное определение проводят методом абсолютной калибровки с систематическим контролем калибровочного коэффициента.
Нижний предел определения - 0,1 мкг/кг.
9. ОТБОР ПРОБ
Отбор проб производят в соответствии с п. 2 настоящих методических указаний.
10. АППАРАТУРА, МАТЕРИАЛЫ И РЕАКТИВЫ
К указанным в п. 3 настоящих методических указаний:
Газовый хроматограф любой марки
Детектор хемилюминисцентный - анализатор термической энергии ТЕА-502 фирмы «Termo Electron Corporation» (США)
Самописец - любой марки
Колонка газохроматографическая стеклянная длиной 3 м и диаметром 2 мм, заполненная 15 % Carbowax-20M, нанесенном на Chromaton N-AW-DMCS (80-100 меш);
Азот газообразный (осч или поверочный нулевой газ) по ГОСТ 9293-74
Кислород газообразный по ГОСТ 5583-78
Аргон газообразный по ГОСТ 10157-90
Азот жидкий
N-нитрозодиметиламин (НДМА) фирмы «Ferrak» (ФРГ)
N-нитрозодиэтиламин (НДЭА) фирмы «Serva» (ФРГ);
N-нитрозодипропиламин (НДПА) фирм» «Serva» (ФРГ)
N-нитрозодибутиламин (НДБА) фирмы «Serva» (ФРГ)
N-нитрозопиперидин (НПиП) фирмы «Serva» (ФРГ)
Н-нитрозопирролидин (НПир) фирмы «Fluka» (Швейцария)
Гексановый раствор НДПА (0,2 мкг/мл) - внутренний стандарт
Гексановый раствор смеси стандартных НА (НДМА, НДЭА, НДБА, НПиП, НПир) по 0,2 мкг каждого НА в 1 мл раствора
11. ТРЕБОВАНИЯ К ТЕХНИКЕ БЕЗОПАСНОСТИ
Требования к технике безопасности и чистоте растворителей и реактивов должны соблюдаться в соответствии с п. 4 и п. 5.1.6. настоящих методических указаний.
12. ПОДГОТОВКА К ИСПЫТАНИЮ И ПРОВЕДЕНИЕ АНАЛИЗА
НА выделяют из анализируемых образцов продовольственного сырья, пищевых продуктов и кормов путем перегонки с паром или вакуумной перегонкой с последующим извлечением НА хлористым метиленом из водного дистиллята и концентрированием хлорметиленового экстракта до 1 мл в соответствии с п. 6.1. и п. 6.2. настоящих методических указаний. Анализ НА осуществляют газохроматографически без дополнительной обработки хлорметиленового или гексанового раствора при следующих условиях: колонка стеклянная (длина 3 м с диаметром 2 мм), заполненная 15 % Carbowax-20M, нанесенном на Chromaton N-AW-DMCS (80 - 100 меш), газноситель - азот (осч или ПНГ) или аргон, скорость - 20 мл/мин, температура колонки - 125 °С (изотермический режим); температура испарителя - 220 °С, температура каталитической печи - 450 °С, давление азота - 0,5 aтм (или расход - 40 мл/мин), давление кислорода - 0,6 атм., температура охлаждающей ловушки - 110 - 130 °С, объем вводимой в хроматограф пробы - 1 - 10 мкл.
Примечания:
1. Может быть использована другая по размеру набивная колонка с содержанием Carbowax, например, 10 %.
2. Измерения целесообразно проводить при одном значении аттенюатора, в обратном случае необходимо убедиться в линейности зависимости показаний аттенюатора и величины сигнала.
Записывают хроматограмму эталонной смеси НА и раствора внутреннего стандарта в начале и в конце каждой серии анализов. Идентификацию НА в пробе осуществляют по временам удерживания стандартов. Количество НА в пробе оценивают сравнением величины аналитических сигналов, полученных при анализе образцов и стандартных растворов.
Оценивается также полнота извлечения НА из продовольственного сырья, пищевых продуктов и кормов по внутреннему стандарту (НДПА вносимому в продукт перед выделением НА).
Содержание НА в пробе рассчитывается по формуле:
![]()
где С - содержание исследуемого НА в образце, мкг/кг,
S1 - площадь под интегральной кривой, полученной при анализе образца мм2,
S2 - площадь под интегральной кривой соответствующего стандартного НА, мм2,
V1 - объем пробы, введенной в хроматограф, мкл,
V2 - объем анализируемого экстракта, мкл,
n - количество стандартного НА, введенного в хроматограф, нг,
m - масса пробы, взятой на анализ, г,
А - степень извлечения внутреннего стандарта, %.
Применяемая методика дает возможность извлекать из анализируемых образцов летучие НА с выходом 70 - 95 %.
Нижний предел определения 1´10-13 мол/мкл.
Метод дает возможность определять 0,1 мкг НА/кг продукта и выше. В интервале концентраций 1´10-10 - 1´10-13 мол/мкл аналитический сигнал прямо пропорционален количеству введенного в хроматограф НА. Относительное стандартное отклонение - 15 - 20 %.
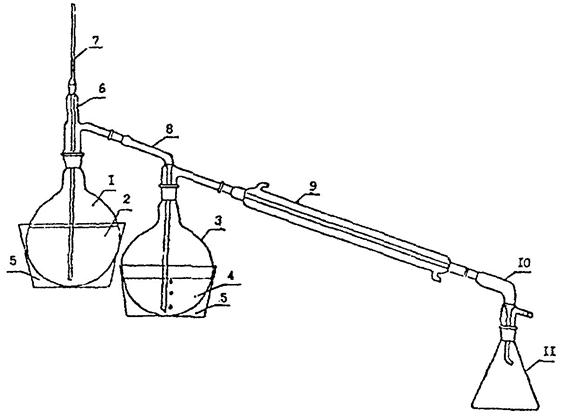
1 - Паровик
2 - Дистиллированная вода
3 - Круглодонная колба
4 - Гологенат пищевого продукта
5 - Баня с глицерином
6 - Насадка Бюрца
7 - Трубка на шлифе
8 - Насадка-барбатер
9 - Холодильник Либиха
10 - Алонж
11 - Приемник
Рис. 1

Рис. 2 СХЕМА ВАКУУМНОЙ УСТАНОВКИ.
ИНФОРМАЦИОННЫЕ ДАННЫЕ
1. Методические указания разработаны:
Институтом питания РАМН (член-корреспондент РАМН, профессор Тутельян В.А., доктор биологических наук Жукова Г.Ф., кандидат медицинских наук Хотимченко С.А.),
Институтом биологической и медицинской химии РАМН (кандидат химических наук Грачева И.Н.),
Онкологическим научным центром РАМН (доктор биологических наук, профессор Хесина А.Я., кандидат биологических наук Кривошеева Л.В., Сокольская Н.Н.),
ВНИРО (кандидат технических наук Головин А.И., Волкова И.М.).
2. Метод идентификации и количественного определения летучих канцерогенных N-нитрозаминов в продовольственном сырье и пищевых продуктах № 4046-85 от 22 ноября 1985 г. утратил силу.
СОДЕРЖАНИЕ
|
3. Аппаратура, материалы, реактивы.. 2 4. Требования техники безопасности при проведении испытаний. 3 10. Аппаратура, материалы и реактивы.. 8 |