|
МЕЖГОСУДАРСТВЕННЫЙ
СОВЕТ ПО СТАНДАРТИЗАЦИИ, МЕТРОЛОГИИ И СЕРТИФИКАЦИИ INTERSTATE COUNCIL
FOR STANDARDIZATION, METROLOGY AND CERTIFICATION |
|
|
МЕЖГОСУДАРСТВЕННЫЙ |
ГОСТ |
МЕДЬ
Методы анализа
|
|
Москва |
Предисловие
Цели, основные принципы и основной порядок проведения работ по межгосударственной стандартизации установлены ГОСТ 1.0-92 «Межгосударственная система стандартизации. Основные положения» и ГОСТ 1.2-97 «Межгосударственная система стандартизации, Стандарты межгосударственные, правила и рекомендации по межгосударственной стандартизации. Порядок разработки, принятия, применения, обновления и отмены»
Сведения о стандарте
1 РАЗРАБОТАН Техническим комитетом по стандартизации ТК 368 «Медь»
2 ВНЕСЕН Техническим секретариатом Межгосударственного совета по стандартизации, метрологии и сертификации
3 ПРИНЯТ Межгосударственным советом по стандартизации, метрологии и сертификации (протокол № 35 от 11 июня 2009 г.)
За принятие стандарта проголосовали:
|
Краткое наименование страны по МК (ИСО 3166) 004-97 |
Код страны по МК (ИСО 3166) 004-97 |
Сокращенное наименование национального органа по стандартизации |
|
Азербайджан |
AZ |
Азстандарт |
|
Беларусь |
BY |
Госстандарт Республики Беларусь |
|
Казахстан |
KZ |
Госстандарт Республики Казахстан |
|
Кыргызстан |
KG |
Кыргызстандарт |
|
Молдова |
MD |
Молдова-Стандарт |
|
Российская Федерация |
RU |
Федеральное агентство по техническому регулированию и метрологии |
|
Таджикистан |
TJ |
Таджикстандарт |
|
Узбекистан |
UZ |
Узстандарт |
|
Украина |
UA |
Госпотребстандарт Украины |
4 В настоящем стандарте учтены основные нормативные положения следующих международных стандартов:
- ИСО 5956:1984 «Медь и медные сплавы. Определение содержания сурьмы. Спектрометрический метод с родамином В» (ISO 5956:1984 «Copper and copper alloys - Determination of antimony content - Rhodamine В spectrometry method», NEQ);
- ИСО 5959:1984 «Медь и медные сплавы. Определение содержания висмута. Спектрометрический метод с применением диэтилдитиокарбамата» (ISO. 5959:1984 «Copper and copper alloys-Determination of bismuth content- Diethyldithiocarbamate spectrometric method», NEQ)
5 Приказом Федерального агентства по техническому регулированию и метрологии от 10 сентября 2009 г. № 322-ст межгосударственный стандарт ГОСТ 31382-2009 введен в действие в качестве национального стандарта Российской Федерации с 1 апреля 2010 г.
6 ВЗАМЕН ГОСТ 13938.1-78 - ГОСТ 13938.10-78, ГОСТ 13938.12-78, ГОСТ 13938.15-88, ГОСТ 9717.1-82, ГОСТ 27981.0-88, ГОСТ 27981.3-88, ГОСТ 27981.4-88
Информация о введении в действие (прекращении действия) настоящего стандарта публикуется в указателе «Национальные стандарты».
Информация об изменениях к настоящему стандарту публикуется в указателе «Национальные стандарты», а текст изменений - в информационных указателях «Национальные стандарты». В случае пересмотра или отмены настоящего стандарта соответствующая информация будет опубликована в информационном указателе «Национальные стандарты»
СОДЕРЖАНИЕ
МЕЖГОСУДАРСТВЕННЫЙ СТАНДАРТ
|
МЕДЬ Copper. Methods of analysis |
Дата введения – 2010-04-01
1 Область применения
Настоящий стандарт распространяется на медь по ГОСТ 859 и устанавливает общие требования к методам анализа/измерений меди, требования безопасности при проведении анализа/измерений, методы выполнения анализа/измерений массовых долей меди и примесей в ней.
2 Нормативные ссылки
В настоящем стандарте использованы нормативные ссылки на следующие межгосударственные стандарты:
ГОСТ 8.315-97 Государственная система обеспечения единства измерений. Стандартные образцы состава и свойств веществ и материалов. Основные положения
ГОСТ 12.0.004-90 Система стандартов безопасности труда. Организация обучения безопасности труда. Общие положения
ГОСТ 12.1.004-91 Система стандартов безопасности труда. Пожарная безопасность. Общие требования
ГОСТ 12.1.005-88 Система стандартов безопасности труда. Общие санитарно-гигиенические требования к воздуху рабочей зоны
ГОСТ 12.1.007-76 Система стандартов безопасности труда. Вредные вещества. Классификация и общие требования безопасности
ГОСТ 12.1.010-76 Система стандартов безопасности труда. Взрывобезопасность. Общие требования
ГОСТ 12.1.016-79 Система стандартов безопасности труда. Воздух рабочей зоны. Требования к методикам измерения концентраций вредных веществ
ГОСТ 12.1.030-81 Система стандартов безопасности труда. Электробезопасность. Защитное заземление, зануление
ГОСТ 12.2.007.0-75 Система стандартов безопасности труда. Изделия электротехнические. Общие требования безопасности
ГОСТ 12.4.009-83 Система стандартов безопасности труда. Пожарная техника для защиты объектов. Основные виды. Размещение и обслуживание
ГОСТ 12.4.021-75 Система стандартов безопасности труда. Системы вентиляционные. Общие требования
ГОСТ 61-75 Реактивы. Кислота уксусная. Технические условия
ГОСТ 83-79 Реактивы, Натрий углекислый. Технические условия
ГОСТ 123-2008 Кобальт. Технические условия
ГОСТ 193-79 (ИСО 431-81) Слитки медные. Технические условия
ГОСТ 199-78 Реактивы. Натрий уксуснокислый 3-водный. Технические условия
ГОСТ 200-76 Реактивы. Натрий фосфорноватистокислый 1-водный. Технические условия
ГОСТ 334-73 Бумага масштабно-координатная. Технические условия
ГОСТ 546-2001 Катоды медные. Технические условия
ГОСТ 804-93 Магний первичный в чушках. Технические условия
ГОСТ 849-2008 Никель первичный. Технические условия
ГОСТ 859-2001 Медь. Марки
ГОСТ 860-75 Олово. Технические условия
ГОСТ 1089-82 Сурьма. Технические условия
ГОСТ 1277-75 Реактивы. Серебро азотнокислое. Технические условия
ГОСТ 1467-93 Кадмий. Технические условия
ГОСТ 1770-74 (ИСО 1042-83, ИСО 4788-80) Посуда мерная лабораторная стеклянная. Цилиндры, мензурки, колбы, пробирки. Общие технические условия
ГОСТ 2062-77 Реактивы. Кислота бромисто-водородная. Технические условия
ГОСТ 3117-78 Реактивы. Аммоний уксуснокислый. Технические условия
ГОСТ 3118-77 Реактивы. Кислота соляная. Технические условия
ГОСТ 3640-94 Цинк. Технические условия
ГОСТ 3652-69 Реактивы. Кислота лимонная моногидрат и безводная. Технические условия
ГОСТ 3760-79 Реактивы. Аммиак водный. Технические условия
ГОСТ 3765-78 Реактивы. Аммоний молибденовокислый. Технические условия
ГОСТ 3773-72 Реактивы. Аммоний хлористый. Технические условия
ГОСТ 3778-98 Свинец. Технические условия
ГОСТ 4109-79 Реактивы. Бром. Технические условия
ГОСТ 4147-74 Реактивы. Железо (III) хлорид 6-водный. Технические условия
ГОСТ 4159-79 Реактивы. Йод. Технические условия
ГОСТ 4165-78 Реактивы. Медь (II) сернокислая 5-водная. Технические условия
ГОСТ 4166-76 Реактивы. Натрий сернокислый. Технические условия
ГОСТ 4198-75 Реактивы. Калий фосфорнокислый однозамещенный. Технические условия
ГОСТ 4201-79 Реактивы. Натрий углекислый кислый. Технические условия
ГОСТ 4204-77 Реактивы. Кислота серная. Технические условия
ГОСТ 4208-72 Реактивы. Соль закиси железа и аммония двойная сернокислая (соль Мора). Технические условия
ГОСТ 4212-76 Реактивы. Приготовление растворов для колориметрического и нефелометрического анализа
ГОСТ 4220-75 Реактивы. Калий двухромовокислый. Технические условия
ГОСТ 4232-74 Реактивы. Калий йодистый. Технические условия
ГОСТ 4233-77 Реактивы. Натрий хлористый. Технические условия
ГОСТ 4236-77 Реактивы. Свинец (II) азотнокислый. Технические условия
ГОСТ 4328-77 Реактивы. Натрия гидроокись. Технические условия
ГОСТ 4329-77 Реактивы. Квасцы алюмокалиевые. Технические условия
ГОСТ 4459-75 Реактивы. Калий хромовокислый. Технические условия
ГОСТ 4461-77 Реактивы. Кислота азотная. Технические условия
ГОСТ 4465-74 Реактивы. Никель (II) сернокислый 7-водный. Технические условия
ГОСТ 4478-78 Реактивы. Кислота сульфосалициловая 2-водная. Технические условия
ГОСТ 4517-87 Реактивы. Методы приготовления вспомогательных реактивов и растворов, применяемых при анализе
ГОСТ 4520-78 Реактивы. Ртуть (II) азотнокислая 1-водная. Технические условия
ГОСТ 4960-2009 Порошок медный электролитический. Технические условия
ГОСТ 5456-79 Реактивы. Гидроксиламина гидрохлорид. Технические условия
ГОСТ 5457-75 Ацетилен растворенный и газообразный технический. Технические условия
ГОСТ 5556-81 Вата медицинская гигроскопическая. Технические условия
ГОСТ 5583-78 (ИСО 2046-73) Кислород газообразный технический и медицинский. Технические условия
ГОСТ 5644-75 Сульфит натрия безводный. Технические условия
ГОСТ 5789-78 Реактивы. Толуол. Технические условия
ГОСТ 5817-77 Реактивы. Кислота винная. Технические условия
ГОСТ 5828-77 Реактивы. Диметилглиоксим. Технические условия
ГОСТ 5845-79 Реактивы. Калий-натрий виннокислый 4-водный. Технические условия
ГОСТ 5905-2004 (ИСО 10387:1994) Хром металлический. Технические требования и условия поставки
ГОСТ 6008-90 Марганец металлический и марганец азотированный. Технические условия
ГОСТ 6344-73 Реактивы. Тиомочевина. Технические условия
ГОСТ 6563-75 Изделия технические из благородных металлов и сплавов. Технические условия
ГОСТ 6709-72 Вода дистиллированная. Технические условия
ГОСТ 6836-2002 Серебро и сплавы на его основе. Марки
ГОСТ 8655-75 Фосфор красный технический. Технические условия
ГОСТ 8677-76 Реактивы. Кальция оксид. Технические условия
ГОСТ 8864-71 Реактивы. Натрия N, N-диэтилдитиокарбамат 3-водный. Технические условия
ГОСТ 9147-80 Посуда и оборудование лабораторные фарфоровые. Технические условия
ГОСТ 9336-75 Реактивы. Аммоний ванадиевокислый мета. Технические условия
ГОСТ 9849-86 Порошок железный. Технические условия
ГОСТ 10157-79 Аргон газообразный и жидкий. Технические условия
ГОСТ 10163-76 Реактивы. Крахмал растворимый. Технические условия
ГОСТ 10298-79 Селен технический. Технические условия
ГОСТ 10652-73 Реактивы. Соль динатриевая этилендиамин-N, N, N', N'-тетрауксусной кислоты, 2-водная (трилон Б). Технические условия
ГОСТ 10727-91 Нити и волокна стеклянные однонаправленные. Технические условия
ГОСТ 10928-90 Висмут. Технические условия
ГОСТ 10929-76 Реактивы. Водорода пероксид. Технические условия
ГОСТ 11069-2001 Алюминий первичный. Марки
ГОСТ 11125-84 Кислота азотная особой чистоты. Технические условия
ГОСТ 11293-89 Желатин. Технические условия
ГОСТ 11773-76 Реактивы. Натрий фосфорнокислый двузамещенный. Технические условия
ГОСТ 12026-76 Бумага фильтровальная лабораторная. Технические условия
ГОСТ 14261-77 Кислота соляная особой чистоты. Технические условия
ГОСТ 14262-78 Кислота серная особой чистоты. Технические условия
ГОСТ 17022-81 Графит. Типы, марки и общие технические требования
ГОСТ 18300-87 Спирт этиловый ректификованный технический. Технические условия
ГОСТ 19908-90 Тигли, чаши, стаканы, колбы, воронки, пробирки и наконечники из прозрачного кварцевого стекла. Общие технические условия
ГОСТ 20015-88 Хлороформ. Технические условия
ГОСТ 20288-74 Реактивы. Углерод четыреххлористый. Технические условия
ГОСТ 20298-74 Смолы ионообменные. Катиониты. Технические условия
ГОСТ 20301-74 Смолы ионообменные. Аниониты. Технические условия
ГОСТ 20448-90 Газы углеводородные сжиженные топливные для коммунально-бытового потребления. Технические условия
ГОСТ 20478-75 Реактивы. Аммоний надсернокислый. Технические условия
ГОСТ 20490-75 Реактивы. Калий марганцовокислый. Технические условия
ГОСТ 21241-89 Пинцеты медицинские. Общие технические требования и методы испытаний
ГОСТ 22180-76 Реактивы. Кислота щавелевая. Технические условия
ГОСТ 22861-93 Свинец высокой чистоты. Технические условия
ГОСТ 22867-77 Реактивы. Аммоний азотнокислый. Технические условия
ГОСТ 24104-2001 Весы лабораторные. Общие технические требования
ГОСТ 24231-80 Цветные металлы и сплавы. Общие требования к отбору и подготовке проб для химического анализа
ГОСТ 24363-80 Реактивы. Калия гидроокись. Технические условия
ГОСТ 25086-87 Цветные металлы и их сплавы. Общие требования к методам анализа
ГОСТ 25336-82 Посуда и оборудование лабораторные стеклянные. Типы, основные параметры и размеры
ГОСТ 25644-96 Средства моющие синтетические порошкообразные. Общие технические требования
ГОСТ 25794.1-83 Реактивы. Методы приготовления титрованных растворов для кислотно-основного титрования
ГОСТ 27025-86 Реактивы, Общие указания по проведению испытаний
ГОСТ 27067-86 Реактивы. Аммоний роданистый. Технические условия
ГОСТ 27068-86 Реактивы. Натрий серноватистокислый (натрия тиосульфат) 5-водный. Технические условия
ГОСТ 29169-91 (ИСО 648-77) Посуда лабораторная стеклянная. Пипетки с одной отметкой
ГОСТ 29227-91 (ИСО 835-1-81) Посуда лабораторная стеклянная. Пипетки градуированные. Часть 1. Общие требования
ГОСТ 29251-91 (ИСО 385-1-84) Посуда лабораторная стеклянная. Бюретки. Часть 1. Общие требования
СТ СЭВ 543-77 Числа. Правила записи и округления
Примечание - При пользовании настоящим стандартом целесообразно проверить действие ссылочных стандартов по указателю «Национальные стандарты», составленному по состоянию на 1 января текущего года, и по соответствующим информационным указателям, опубликованным в текущем году. Если ссылочный стандарт заменен (изменен), то при пользовании настоящим стандартом следует руководствоваться заменяющим (измененным) стандартом. Если ссылочный стандарт отменен без замены, то положение, в котором дана ссылка на него, применяется в части, не затрагивающей эту ссылку.
3 Общие требования
3.1 Общие требования к методам анализа/измерений - по ГОСТ 25086.
3.2 Общие требования к средствам измерений, вспомогательным устройствам, материалам, реактивам, растворам - по ГОСТ 25086.
3.3 Приготовление растворов химических реактивов - в соответствии с ГОСТ 4212, ГОСТ 4517, ГОСТ 25794.1 и ГОСТ 27025.
3.4 Допускается применение других средств измерений, вспомогательных устройств, материалов, реактивов, обеспечивающих проведение анализа/измерений с установленной погрешностью.
3.5 Отбор и подготовку проб меди к анализу/измерениям осуществляют по ГОСТ 193, ГОСТ 546 или ГОСТ 24231.
3.6 Для взвешивания применяют лабораторные весы по ГОСТ 24104. В методике анализа/измерений должен быть указан класс точности весов.
3.7 Массовую долю меди определяют параллельно в трех навесках, примесей - по количеству параллельных определений, число которых указывается в конкретном методе анализа/измерений, но не менее двух. Одновременно с проведением анализа/измерений в тех же условиях проводят контрольный опыт для внесения соответствующей поправки в результаты анализа/измерений. При определении меди проводят два контрольных опыта. При определении примесей число параллельных определений при контрольном опыте должно соответствовать числу параллельных определений, указанному в методе анализа/измерений.
3.8 Для прокаливания и сплавления применяют муфельные лабораторные печи, обеспечивающие нагревание до температуры 1000°С. Для высушивания применяют лабораторные сушильные печи, обеспечивающие нагревание до температуры 250°С. Для растворения и выпаривания растворов применяют электрические плиты с закрытой спиралью, обеспечивающие нагревание до температуры 350°С.
3.9 Для измерения промежутков времени менее 5 мин применяют песочные часы и секундомеры, более 5 мин - таймеры или часы любого типа.
3.10 Термины, касающиеся степени нагрева воды (раствора) и продолжительности операций, - по ГОСТ 27025.
3.11 Для приготовления растворов с известной массовой концентрацией используют металлы и их соединения с массовой долей основного компонента не менее 99,9%, если в методике выполнения измерений не предусмотрено иное. Способ приготовления растворов - по ГОСТ 4212 или по настоящему стандарту.
3.12 Взвешивание анализируемого вещества, вещества для приготовления растворов с известной концентрацией металлов и осадков в гравиметрическом анализе проводится, если это специально не оговорено в методике анализа, на весах специального класса точности по ГОСТ 24104.
3.13 Проверка приемлемости результатов анализа/измерений и установление окончательного результата - в соответствии со стандартами [1], [2].
3.14 Контроль точности результатов анализа/измерений
Контроль точности результатов анализа/измерений проводят в соответствии с рекомендациями [3]:
а) сопоставлением результата контрольной процедуры с нормативом контроля. Результат контрольной процедуры Кк рассчитывают по формуле
|
|
(1) |
где ![]() - результат
анализа/измерения стандартного образца (СО);
- результат
анализа/измерения стандартного образца (СО);
С - аттестованное значение СО.
Норматив контроля К рассчитывают по формуле
|
К = ∆n, |
(2) |
где ±Δn - значение характеристики погрешности результата анализа/измерения при реализации в конкретной лаборатории, соответствующее аттестованному значению СО.
Если при проведении контроля применяют СО, которые не использовались при установлении показателя точности результатов анализа/измерения, и в случае превышения погрешности СО одной трети погрешности методики анализа/измерения, норматив контроля точности рассчитывают по формуле
|
|
(3) |
где Δатт - характеристика погрешности аттестованного значения измеряемого элемента в СО.
б) используя СО состава, утвержденные в соответствии с ГОСТ 8.315. Периодичность измерения состава СО - в соответствии с руководством по обеспечению качества аналитических работ, действующем на предприятии.
Массовую долю определяемого компонента в СО находят путем параллельных измерений, установленных конкретным методом анализа/измерений.
Для контроля стабильности результатов анализа/измерений рекомендуется использовать контрольные карты (КК) Шухарта по стандартам [2] (раздел 6) и [4].
Алгоритмы оценки стабильности результатов анализа/измерений - в соответствии с руководством по обеспечению качества аналитических работ, действующим на предприятии, с учетом требований стандарта [2] (раздел 6).
При отсутствии СО допускается контроль точности результатов анализа/измерений проводить по ГОСТ 25086 с использованием метода добавок или аттестованных смесей по рекомендациям [5].
3.15 Оформление результатов анализа/измерений
Результаты анализа/измерений представляют в виде Х ±∆ (при доверительной вероятности Р = 0,95),
где X - результат анализа/измерений, %;
∆ - погрешность результатов анализа/измерений, %.
Значения ∆ приведены в конкретной методике анализа/измерений.
Примечание - В случае, когда за окончательный результат анализа/измерений принимают медиану, то результат представляют без указания границ погрешности.
3.16 Допускается построение градуировочных графиков и расчет результатов анализа/измерений проводить с использованием программного обеспечения используемых средств измерений. В этом случае программное обеспечение должно быть сертифицировано.
3.17 Округление результатов анализа/измерений проводят в соответствии с требованиями СТ СЭВ 543.
4 Требования безопасности
4.1 Подготовка проб к анализу и проведение анализа (растворение в кислотах, щелочах и пр.) и все операции химического анализа, связанные с выделением ядовитых паров или газов, следует выполнять в вытяжных шкафах или боксах, оборудованных местным отсасывающим устройством по ГОСТ 12.4.021.
4.2 Лабораторные помещения должны быть оборудованы вентиляционными системами по ГОСТ 12.4.021.
4.3 При выполнении анализа меди в воздух рабочей зоны могут выделяться вредные вещества, предельно допустимые концентрации (ПДК) их в воздухе рабочей зоны должны соответствовать ГОСТ 12.1.005 и гигиеническим нормативам [6].
4.4 Контроль за содержанием вредных веществ в воздухе рабочей зоны следует осуществлять в соответствии с требованиями ГОСТ 12.1.005, ГОСТ 12.1.007 и ГОСТ 12.1.016.
4.5 Лабораторные помещения, в которых выполняется работа по химическому анализу исследуемого материала, должны соответствовать требованиям пожарной безопасности по ГОСТ 12.1.004 и правилам пожарной безопасности [7]. Средства и способы пожаротушения следует применять по ГОСТ 12.4.009 в зависимости от источника возникновения и характера пожара.
4.6 При работе с горючими и взрывоопасными газами следует соблюдать требования ГОСТ 12.1.010, ГОСТ 12.1.004. При использовании газов в баллонах следует соблюдать требования правил [8].
4.7 Электротехнические контрольно-измерительные приборы и лабораторное оборудование и условия их эксплуатации должны соответствовать требованиям ГОСТ 12.2.007.0, ГОСТ 12.1.030 и стандарта [9]. Заземление должно соответствовать требованиям правил [10].
4.8 Организация обучения безопасности труда и проверка знаний работающих требований безопасности труда - по ГОСТ 12.0.004.
4.9 Персонал лаборатории должен быть обеспечен специальной одеждой, специальной обувью и другими средствами индивидуальной защиты в соответствии с правилами [11].
4.10 Персонал лаборатории должен быть обеспечен бытовыми помещениями по группе производственных процессов IIIa в соответствии со строительными нормами и правилами [12].
5 Методы определения массовой доли меди
5.1 Область применения
В настоящем разделе установлены электрогравиметрический и расчетный методы определения массовой доли меди.
5.2 Требования к погрешности анализа
Погрешность результатов анализа (при массовой доле меди 99,00% и выше) для доверительной вероятности Р = 0,95 не должна превышать ±0,10%.
5.3 Средства измерений, вспомогательные устройства, материалы, растворы
При выполнении анализа применяют следующие средства измерений, вспомогательные устройства:
- электроды из платины сетчатые по ГОСТ 6563;
- установку для электролиза с амперметром, вольтметром, реостатом, обеспечивающую проведение электролиза при перемешивании при плотности тока от 2 до 3 А/дм3 и напряжении от 2,2 до 2,5 В;
- фотометр фотоэлектрический или спектрофотометр со всеми принадлежностями;
- спектрофотометр атомно-абсорбционный, включающий источник излучения на медь, горелку для пламени ацетилен-воздух и распылительную систему;
- компрессор воздушный;
- центрифугу со всеми принадлежностями;
- шкаф сушильный с терморегулятором;
- весы лабораторные специального класса точности по ГОСТ 24104;
- пипетки не ниже 2-го класса точности по ГОСТ 29169 и ГОСТ 29227;
- стаканы В-1-50 ТХС; В-1-100 ТХС; В-1-250 ТС по ГОСТ 25336;
- колбы мерные 2-25-2, 2-100-2, 2-200-2, 2-250-2, 2-1000-2 по ГОСТ 1770;
- воронку ВД-1-100 ХС по ГОСТ 25336;
- эксикатор 2-190 по ГОСТ 25336.
При выполнении анализа применяют следующие материалы, растворы:
- ацетилен по ГОСТ 5457;
- кислоту азотную по ГОСТ 4461;
- кислоту серную по ГОСТ 4204 и разбавленную 1:1;
- аммоний азотнокислый по ГОСТ 22867;
- смесь для растворения;
- кислоту лимонную по ГОСТ 3652;
- аммиак водный по ГОСТ 3760, разбавленный 1:4;
- соль динатриевую этилендиамин-N, N, N', N'-тетрауксусной кислоты, двуводную (трилон Б) по ГОСТ 10652, 0,1 М раствор;
- купризон, бис-(циклогексанон) оксалилдигидразон, раствор массовой концентрации 2,5 г/дм3;
- натрий сернокислый безводный по ГОСТ 4166;
- фенолфталеин (индикатор) по [13], спиртовый раствор массовой концентрации 1 г/дм3;
- углерод четыреххлористый по ГОСТ 20288;
- спирт этиловый ректификованный по ГОСТ 18300;
- медь по ГОСТ 859;
- растворы меди известной концентрации;
- бумагу индикаторную универсальную по техническим условиям [14];
- хлороформ по ГОСТ 20015;
- диэтилдитиокарбамат свинца (II) по [15], раствор массовой концентрации 0,2 г/дм3 в хлороформе.
5.4 Метод анализа
Метод основан на электролитическом выделении меди из раствора серной и азотной кислот в присутствии солей аммония на платиновых сетчатых электродах при плотности тока от 2 до 3 А/дм2 и напряжении от 2,2 до 2,5 В.
Медь, оставшуюся в электролите, определяют атомно-абсорбционным или фотометрическим методом. В случае разногласий при оценке массовой доли меди используют фотометрический метод, основанный на образовании окрашенного комплексного соединения меди с купризоном или диэтилдитиокарбаматом свинца.
При массовой доле меди от 99,00% до 99,90% медь в сумме с серебром определяют электролитически.
Массовую долю меди свыше 99,90% определяют по разности, вычитая сумму определенных примесей из 100%.
5.5 Подготовка к выполнению анализа
5.5.1 При приготовлении смеси для растворения навеску 500 г азотнокислого аммония растворяют в 500 см3 воды, прибавляют 500 см3 азотной кислоты, 200 см3 серной кислоты и доливают водой до 2000 см3.
5.5.2 При приготовлении раствора лимоннокислого аммония навеску 150 г лимонной кислоты растворяют в 400 см3 воды, прибавляют 200 см3 раствора аммиака, охлаждают, доливают до 1000 см3 водой и перемешивают.
5.5.3 При приготовлении 0,1 М раствора трилона Б навеску 37,2 г трилона Б растворяют в 800 см3 воды, доливают водой до 1000 см3 и хорошо перемешивают.
5.5.4 При приготовлении раствора купризона массовой концентрации 2,5 г/дм3 навеску 2,5 г купризона растворяют при перемешивании в 900 см3 воды при температуре от 70°С до 800С. После охлаждения раствор фильтруют в сосуд из темного стекла, доливают водой до 1000 см3, перемешивают и хранят в этом сосуде. Раствор годен к применению в течение 10 суток.
5.5.5 Для построения градуировочных графиков готовят растворы меди известной концентрации.
При приготовлении раствора А массовой концентрации меди 0,5 мг/см3 навеску 0,5000 г меди растворяют в 20 см3 смеси для растворения и при нагревании удаляют окислы азота. Раствор охлаждают, разбавляют водой до 100 см3 и помещают в мерную колбу вместимостью 1000 см3, доливают водой до метки и перемешивают.
При приготовлении раствора Б массовой концентрации меди 0,01 мг/см3 20 см3 раствора А помещают в мерную колбу вместимостью 1000 см3, прибавляют 5 см3 серной кислоты, разбавленной 1:1, доливают до 1000 см3 водой и перемешивают.
5.5.6 Для приготовления раствора диэтилдитиокарбамата свинца (II) массовой концентрации 0,2 г/дм3 в хлороформе навеску 0,2 г диэтилдитиокарбамата свинца (II) помещают в мерную колбу вместимостью 1000 см3, добавляют от 100 до 200 см3 хлороформа и перемешивают до растворения навески, доливают до метки хлороформом и снова перемешивают. Раствор хранят в склянке из темного стекла в темном месте.
5.5.7 Построение градуировочных графиков
Отбирают 0; 2,0; 4,0; 6,0; 8,0 и 10,0 см3 раствора Б и помещают в мерные колбы вместимостью 100 см3 каждая, что соответствует 0; 20; 40; 60; 80 и 100 мкг меди. Прибавляют 4 см3 смеси для растворения, 50 см3 воды, 10 см3 раствора лимоннокислого аммония, 2 капли раствора фенолфталеина, раствор аммиака, разбавленный 1:4, до появления слабо-розовой окраски и 1 см3 избытка, 10 см3 раствора купризона, доливают водой до метки и перемешивают. Величина рН раствора должна быть от 8,5 до 9,0.
Измерение оптической плотности проводят, как указано в 5.6.3.
По найденным значениям оптической плотности и соответствующим им значениям содержания меди строят градуировочный график.
В шесть делительных воронок вместимостью 100 см3 каждая помещают 0; 0,5; 1,0; 2,0; 3,0 и 5,0 см3 раствора Б, что соответствует 0; 5; 10; 20; 30 и 50 мкг меди. Приливают воды до 50 см3 и далее анализ проводят по 5.6.4.
Экстракцию и измерение оптической плотности раствора проводят так, как указано в 5.6.4.
По найденным значениям оптической плотности и соответствующим им значениям содержания меди строят градуировочный график.
В мерные колбы вместимостью 100 см3 каждая отбирают 0; 5,0; 10,0; 15,0 и 20,0 см3 раствора Б, доливают до метки водой и перемешивают. Растворы содержат 0; 0,5; 1,0; 1,5 и 2,0 мкг/см3 меди. Растворы распыляют в пламя и измеряют абсорбцию в пламени при длине волны 324,7 нм.
По найденным значениям оптической плотности и соответствующим значениям содержания меди строят градуировочный график.
5.6 Выполнение анализа
5.6.1 Общие требования к методам анализа и требования безопасности при выполнении работ - в соответствии с разделами 3 и 4.
5.6.2 Электрогравиметрический метод определения меди (при массовой доле от 99,00 % до 99,90 %)
Навеску меди массой от 1,0000 до 2,0000 г помещают на чашку весов, где находится взвешенный платиновый катод, предназначенный для электролиза, и определяют суммарную массу катода и меди. Допускается раздельное взвешивание навески меди и катода, предназначенного для электролиза. Навеску меди переносят в стакан вместимостью 250 см3, прибавляют 40 см3 смеси для растворения и стакан накрывают часовым стеклом. После растворения навески меди раствор осторожно нагревают до удаления окислов азота, разбавляют до 180 см3 водой, нагревают до 40°С и в раствор погружают платиновые электроды. После этого проводят электролиз в течение 2,5 ч при перемешивании раствора при плотности тока от 2 до 3 А/дм3 и напряжении от 2,2 до 2,5 В. Для проверки полноты выделения меди погружают электроды на 5 мм ниже первоначального положения и продолжают электролиз. При отсутствии налета меди на свежепогруженной части катода электролиз считают законченным.
После этого, не выключая ток, платиновые электроды промывают водой, а затем, выключив ток, промывают этиловым спиртом (из расчета 10 см3 спирта на одно определение).
Катод с выделившейся медью сушат при температуре от 100°С до 105°С в течение 5 мин, охлаждают в эксикаторе и взвешивают на весах, на которых взвешивались катод и навеска меди перед анализом.
Электролит с промывными водами (после промывания платиновых катодов) переливают в мерную колбу вместимостью от 200 до 250 см3, доливают водой до метки и перемешивают. Электролит сохраняют для определения никеля.
Медь, оставшуюся в электролите после проведения электролиза, определяют в виде окрашенного соединения с купризоном или диэтилдитиокарбаматом свинца фотометрическим методом так, как описано в 5.6.3 и 5.6.4, или атомно-абсорбционным методом в соответствии с 5.6.5.
5.6.3 Фотометрический метод определения меди в электролите с купризоном
Пипеткой отбирают 50 см3 раствора электролита и помещают в мерную колбу вместимостью 100 см3, добавляют 10 см3 раствора лимоннокислого аммония, 2 капли раствора фенолфталеина и раствор аммиака, разбавленный 1:4, до получения слабо-розовой окраски. Затем прибавляют 1 см3 раствора аммиака, разбавленного 1:4, 10 см3 купризона, доливают до метки водой и перемешивают.
Величина рН раствора должна быть от 8,5 до 9,0, рН раствора проверяют по индикаторной бумаге.
Оптическую плотность раствора измеряют по истечении от 5 до 30 мин при длине волны 600 нм в кювете толщиной поглощающего свет слоя 30 мм. Раствором сравнения при измерении оптической плотности является вода. Одновременно проводят два контрольных опыта со всеми применяемыми реактивами. Среднее значение оптической плотности контрольного опыта вычитают из значения оптической плотности анализируемого раствора.
Массу меди устанавливают по градуировочному графику, построенному, как указано в 5.5.7.1.
5.6.4 Фотометрический метод определения меди в электролите с диэтилдитиокарбаматом свинца
Отбирают аликвотную часть раствора электролита от 5 до 10 см3 и помещают в стакан вместимостью 50 см3, приливают 5 см3 серной кислоты, разбавленной 1:10, и выпаривают до выделения паров серной кислоты.
Раствор охлаждают, приливают от 10 до 20 см3 воды, помещают в делительную воронку вместимостью 100 см3 и разбавляют водой до объема 50 см3. Добавляют 10 см3 раствора диэтилдитиокарбамата свинца и экстрагируют в течение 2 мин. После разделения слоев экстракт сливают в мерную колбу вместимостью 25 см3 (куда предварительно помещают 1 г безводного сернокислого натрия).
Экстракцию повторяют с 10 см3 экстрагента. Органический слой сливают в ту же мерную колбу, разбавляют до метки хлороформом и перемешивают.
Оптическую плотность раствора измеряют при длине волны 413 нм в кювете толщиной поглощающего свет слоя 50 мм. Раствором сравнения при измерении оптической плотности служит четыреххлористый углерод.
Одновременно проводят два контрольных опыта. Для этого помещают в делительную воронку 4 см3 смеси для растворения, доливают до 50 см3 водой и далее поступают, как указано выше. Среднее значение оптической плотности контрольного опыта вычитают из значения оптической плотности анализируемого раствора.
Массу меди устанавливают по градуировочному графику, построенному, как указано в 5.5.7.2.
5.6.5 Атомно-абсорбционный метод определения меди в электролите
Часть раствора электролита помещают в стакан вместимостью 100 см3, предварительно ополоснув его этим раствором. Раствор распыляют в пламя и измеряют абсорбцию в пламени при длине волны 324,7 нм.
Массу меди в растворе устанавливают по градуировочному графику, построенному, как указано в 5.5.7.3.
5.7 Обработка результатов анализа
5.7.1 Массовую долю меди Х, %, при использовании электрогравиметрического и фотометрического методов определения вычисляют по формуле
|
|
(4) |
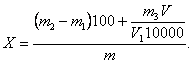
Массовую долю меди Х, %, при использовании электрогравиметрического и атомно-абсорбционного методов определения меди вычисляют по формуле
|
|
(5) |
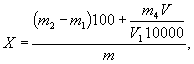
где т2 - масса катода с осажденной медью, г;
m1 - масса катода, г;
m3 - масса меди, найденная по градуировочному графику, мкг;
m4 - масса меди, найденная по градуировочному графику, мкг/см3;
V - объем анализируемого электролита, см3;
V1 - объем аликвотной части электролита, см3;
т - масса навески меди, г.
5.7.2 За результат анализа принимают среднеарифметическое значение трех параллельных определений при условии, что разность между наибольшим и наименьшим результатами в условиях повторяемости при доверительной вероятности Р = 0,95 не превышает значения 0,06%.
Если расхождение между наибольшим и наименьшим результатами параллельных определений превышает значение предела повторяемости, выполняют процедуры, изложенные в стандарте [2] (подпункт 5.2.2.1).
Абсолютное значение допускаемого расхождения между двумя результатами анализа, полученными в разных лабораториях, не должно превышать предела воспроизводимости R = 0,14% для доверительной вероятности Р = 0,95.
5.7.3 Определение меди (при массовой доле ее свыше 99,90%)
5.7.3.1 Массовую долю меди X, %, вычисляют по разности между 100 и суммой всех определяемых примесей по формуле
|
X = 100 – (X1 + X2 + X3 + …+ Xn) |
(6) |
где Х1, Х2, Х3, … Хп - средняя массовая доля определенных в меди примесей, %.
Число значащих цифр зависит от исходных требований, задаваемых в нормативном документе на конкретный вид продукции.
5.7.3.2 Расхождения между результатами двух параллельных определений/измерений примесей в меди не должны превышать пределов повторяемости, приведенных в соответствующих методиках при определении той или иной примеси.
Расхождения между двумя результатами анализа/измерений примесей в меди, полученными в разных лабораториях, не должны превышать значений пределов воспроизводимости, приведенных в соответствующих методиках при определении той или иной примеси.
6 Методы определения массовой доли серы
6.1 Область применения
В настоящем разделе установлены титриметрический метод (при массовой доле серы от 0,0010% до 0,020%) и метод инфракрасной спектрометрии (при массовой доле серы от 0,0002% до 0,050%) определения массовой доли серы в меди.
6.2 Требования к погрешности анализа
Погрешность результатов анализа/измерений массовой доли серы, значения пределов повторяемости и воспроизводимости для доверительной вероятности Р = 0,95 должны соответствовать приведенным в таблицах 1, 2 и 3.
Таблица 1 - Титриметрический метод
В процентах
|
Диапазон массовой доли серы |
Погрешность результатов анализа ±∆ |
Предел |
|
|
повторяемости r (n=2) |
воспроизводимости R |
||
|
От 0,0010 до 0,0030 включ. |
0,0007 |
0,0005 |
0,0010 |
|
Св. 0,003 » 0,006 » |
0,001 |
0,001 |
0,002 |
|
» 0,006 » 0,020 » |
0,003 |
0,002 |
0,004 |
Таблица 2 - Метод инфракрасной спектрометрии в присутствии плавня
В процентах
|
Диапазон массовой доли серы |
Погрешность результатов измерений ±∆ |
Предел |
|
|
повторяемости r (n=2) |
воспроизводимости R |
||
|
От 0,0003 до 0,0005 включ. |
0,0002 |
0,0002 |
0,0003 |
|
Св. 0,0005 » 0,0010 » |
0,0005 |
0,0005 |
0,0007 |
|
» 0,0010 » 0,0030 » |
0,0008 |
0,0008 |
0,0011 |
|
» 0,0030 » 0,0050 » |
0,0011 |
0,0011 |
0,0015 |
|
» 0,0050 » 0,0100 » |
0,0014 |
0,0014 |
0,0018 |
|
» 0,010 » 0,030 » |
0,003 |
0,003 |
0,004 |
|
» 0,030 » 0,050 » |
0,005 |
0,005 |
0,007 |
6.3 Титриметрический метод
6.3.1 Средства измерений, вспомогательные устройства, материалы, растворы
При выполнении анализа применяют следующие средства измерений, вспомогательные устройства:
- весы лабораторные специального класса точности по ГОСТ 24104;
Таблица 3 - Метод инфракрасной спектрометрии без применения плавня
В процентах
|
Диапазон массовой доли серы |
Погрешность результатов измерений ±∆ |
Предел |
|
|
Повторяемости r (n=2) |
воспроизводимости R |
||
|
От 0,0002 до 0,0005 включ. |
0,0001 |
0,0002 |
0,0002 |
|
Св. 0,0005 » 0,0010 » |
0,0002 |
0,0003 |
0,0003 |
|
» 0,0010 » 0,0025 » |
0,0003 |
0,0005 |
0,0005 |
|
» 0,0025 » 0,0050 » |
0,0005 |
0,0006 |
0,0007 |
- колбы мерные 2-25-2; 2-250-2; 2-1000-2 по ГОСТ 1770;
- пипетки не ниже 2-го класса точности по ГОСТ 29227;
- бюретки 1-1-2-25-0,1 по ГОСТ 29251;
- мензурки 50, 100 по ГОСТ 1770;
- колбы Кн-2-250-34 ТХС по ГОСТ 25336;
- стаканы В-1-100 ТХС по ГОСТ 25336;
- печь муфельную с температурой нагрева до 1050°С;
- установку для определения серы согласно рисунку 1;
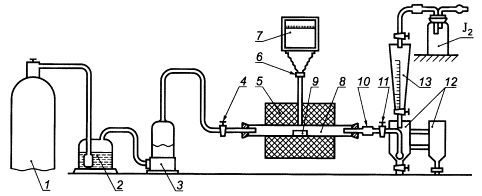
1 - баллон с кислородом, снабженный редукционным вентилем для регулирования скорости поступления кислорода в печь; 2 - промывная склянка, содержащая раствор марганцовокислого калия в растворе гидроксида калия или гидроксида натрия; 3 - склянка, содержащая в нижней части плавленый хлористый кальций и слой стеклянной или обыкновенной ваты, а в верхней части - гидроксид калия или гидроксид натрия; 4 - кран, дающий возможность регулировать подачу очищенного кислорода в трубки для сжигания; 5 - трубчатая печь с силитовыми нагревателями, обеспечивающими нагрев до 1250°С; 6 - термопара; 7 - милливольтметр или потенциометр любого типа; 8 - трубки для сжигания кислорода; 9 - лодочка для сжигания пробы; 10 - очистительный сосуд с кварцевой ватой; 11 - кран перед поглотительным сосудом; 12 - поглотительный сосуд, состоящий из двух одинаковых сосудов, соединенных стеклянными перемычками. Допускается использование двух стеклянных цилиндров высотой по 250 мм из стекла одного цвета (рисунок 2); 13 - бюретка для титрования
Рисунок 1 - Установка для определения серы
- печь трубчатую с силитовыми нагревателями, обеспечивающими нагрев до 1250°С;
- милливольтметр или потенциометр любого типа;
- трубку фарфоровую одноканальную (внешний диаметр - 26 мм, внутренний диаметр - 21 мм, длина - от 850 до 900 мм);
- лодочки фарфоровые ЛС2 по ГОСТ 9147;
- эксикатор 2-190 по ГОСТ 25336, заполненный оксидом кальция, предварительно прокаленным при температуре от 970°С до 1050°С, или хлористым кальцием.
При выполнении анализа применяют следующие материалы, растворы:
- калий двухромовокислый по ГОСТ 4220, дважды перекристаллизованный и высушенный при температуре 170°С, раствор 0,025 н.;
- калий йодистый по ГОСТ 4232, раствор массовой концентрации 50 г/дм3;
- калия гидроксид (калия гидроокись) по ГОСТ 24363, раствор массовой концентрации 400 г/дм3;
- натрия гидроксид (натрия гидроокись) по ГОСТ 4328, раствор массовой концентрации 400 г/дм3;
- калий марганцовокислый по ГОСТ 20490, раствор массовой концентрации 40 г/дм3 в растворе калия гидроксида или натрия гидроксида;
- кальций хлористый по [16], плавленый;
- кислоту серную по ГОСТ 4204, разбавленную 5:100;
- крахмал растворимый по ГОСТ 10163, раствор массовой концентрации 10 г/дм3;
- натрий углекислый безводный по ГОСТ 83;
- натрий серноватистокислый по ГОСТ 27068, раствор 0,025 н.;
- йод по ГОСТ 4159, раствор 0,001 н.;
- кальция оксид по ГОСТ 8677;
- стандартный образец меди, стали (нелегированной) или железа с массовой долей серы от 0,002% до 0,03%.
6.3.2 Метод анализа
Метод основан на сжигании навески меди, содержащей серу, в токе кислорода при температуре 1200°С, поглощении образующейся двуокиси серы водой и титровании сернистой кислоты раствором йода в присутствии крахмала.
6.3.3 Подготовка к выполнению анализа
6.3.3.1 Перед проведением анализа необходимо проверить герметичность установки для определения серы (рисунок 1) и правильность ее сборки.
Для этого соединяют всю установку с баллоном, содержащим кислород, открывают трехходовой кран на воздух, осторожно открывают вентиль баллона, пропускают кислород со скоростью 20 - 30 пузырьков в минуту, переключают трехходовой кран в положение, при котором кислород поступает в печь, и закрывают кран перед поглотительным сосудом. В течение 2 - 3 мин должно прекратиться выделение пузырьков в промывных склянках, после чего необходимо выждать еще от 5 до 7 мин. Если пузырьки больше не выделяются, установку можно считать герметичной.
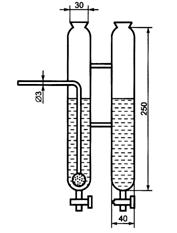
Рисунок 2 - Поглотительный сосуд
6.3.3.2 Перед проведением анализа необходимо проверить при температуре от 1200°С до 1250°С устройство для сжигания на герметичность и наличие летучих восстанавливающих веществ. Для этого в оба сосуда поглотительного устройства наливают по 50 см3 воды и по 10 см3 раствора крахмала, приливают из бюретки несколько капель раствора йода до появления сине-голубой окраски (интенсивность окраски в обоих сосудах должна быть одинаковой). Нагревают печь до температуры от 1100°С до 1250°С и пропускают кислород со скоростью 40 - 50 пузырьков в минуту.
Если через 4 - 5 мин окраска раствора в левом сосуде исчезнет, то это означает, что из трубки выделяются восстанавливающие вещества, реагирующие с йодом. В этом случае, не прекращая тока кислорода, к раствору в левом сосуде приливают еще несколько капель раствора йода и продолжают прибавление раствора йода до тех пор, пока синяя окраска в растворе будет оставаться постоянной и одинаковой по интенсивности с окраской раствора в правом сосуде.
6.3.3.3 Для проведения анализа фарфоровые лодочки предварительно прокаливают при температуре от 850°С до 900°С в течение 1 ч. Прокаленные лодочки помещают в эксикатор. Перед проведением анализа лодочку прокаливают при температуре 1200°С в атмосфере кислорода, проверяют на содержание серы в условиях проведения анализа. Навеску с испытуемым образцом помещают в проверенную лодочку. После проведения испытания лодочку больше не используют.
6.3.3.4 При приготовлении 0,025 н. раствора серноватистокислого натрия (натрия тиосульфата) навеску 6,2 г растворяют в 100 см3 свежепрокипяченной и охлажденной воды, прибавляют 0,2 г безводного углекислого натрия, доливают водой до 1000 см3 и хорошо перемешивают.
Массовую концентрацию раствора серноватистокислого натрия устанавливают на 2 - 3 суток после приготовления раствора.
При установлении массовой концентрации 0,025 н. раствора серноватистокислого натрия 10 см3 серной кислоты, разбавленной 5:100, помещают в коническую колбу вместимостью 250 см3, приливают 10 см3 раствора йодистого калия, 25 см3 0,025 н. раствора двухромовокислого калия. Колбу закрывают пришлифованной пробкой и оставляют в темном месте в течение 8 - 10 мин. Приливают воду до объема от 70 до 80 см3 и титруют выделившийся йод раствором серноватистокислого натрия до светло-желтой окраски, приливают 2 см3 раствора крахмала и продолжают титрование до исчезновения синей окраски.
Массовую концентрацию раствора серноватистокислого натрия NNa, г/см3, вычисляют по формуле
|
|
(7) |
где V - объем раствора серноватистокислого натрия, израсходованный на титрование, см3.
6.3.3.5 При приготовлении 0,001 н. раствора йода навеску 0,127 г йода растворяют в 50 см3 раствора йодистого калия и разбавляют раствор водой до 1 дм3. Раствор хранят в стеклянной посуде из темного стекла.
Титр раствора йода, выраженный в граммах серы, устанавливают по четырем навескам стандартного образца с известным содержанием серы. Сжигание серы в этом случае проводят согласно 6.3.4.
Титр раствора йода по сере T, г, вычисляют по формуле
|
|
(8) |
где С - массовая доля серы в стандартном образце, %;
т - масса стандартного образца, г;
V - объем раствора йода, израсходованный на титрование, см3.
Примечание - При отсутствии стандартного образца массовую концентрацию раствора йода устанавливают по раствору серноватистокислого натрия, массовая концентрация которого установлена по раствору двухромовокислого калия.
При установлении массовой концентрации 0,001 н. раствора йода готовят 0,001 н. раствор серноватистокислого натрия с разбавлением 0,025 н. раствора: отбирают пипеткой 10 см3 0,025 н. раствора серноватистокислого натрия, помещают в мерную колбу вместимостью 250 см3, доливают предварительно прокипяченной и охлажденной водой до метки и перемешивают. Раствор готовят в день применения. В колбу вместимостью 250 см3 наливают от 18 до 20 см3 воды, приливают из бюретки точно отмеренные 20 см3 раствора йода, разбавляют водой до объема от 70 до 80 см3, перемешивают и титруют 0,001 н. раствором серноватистокислого натрия до светло-желтой окраски, затем приливают 2 см3 раствора крахмала и продолжают титрование до исчезновения синей окраски.
Массовую концентрацию раствора йода NJ, г/см3, вычисляют по формуле
|
|
(9) |
где N1 - массовая концентрация раствора серноватистокислого натрия, равная NNa/25, г/см3;
V - объем раствора серноватистокислого натрия, израсходованный на титрование, см3.
Титр раствора йода по сере Т, г, вычисляют по формуле
|
|
(10) |
6.3.4.1 Общие требования к методам анализа и требования безопасности при выполнении анализов - в соответствии с разделами 3 и 4.
6.3.4.2 Массовую долю серы определяют параллельно из двух навесок пробы.
6.3.4.3 Одновременно через все стадии подготовки проб к анализу проводят контрольный опыт на чистоту реактивов.
6.3.4.4 Навеску меди массой 2,0 г (при массовой доле серы до 0,005%) или массой 1,0 г (при массовой доле серы свыше 0,005%) распределяют равномерно по дну предварительно прокаленной лодочки для сжигания.
После этого в трубку печи (в наиболее нагретую зону) помещают лодочку с навеской меди при помощи длинного крючка из стальной проволоки диаметром от 2 до 3 мм. Трубку печи немедленно соединяют с остальными устройствами и сжигают навеску меди. Скорость пропускания кислорода должна поддерживаться такой, чтобы жидкость в поглотительном сосуде (рисунок 2, левая часть) поднималась на дополнительную высоту от 2 до 3 см. Когда поступающие из печи в поглотительный сосуд газы начинают обесцвечивать раствор йода, приливают раствор йода с такой скоростью, чтобы синяя окраска не исчезала во время сжигания навески. Сжигание серы считают законченным, когда окраска раствора в поглотительном растворе остается постоянной и одинаковой по интенсивности с окраской раствора в правой части сосуда для поглощения.
6.3.5 Обработка результатов анализа
6.3.5.1 Массовую долю серы X, %, вычисляют по формуле
|
|
(11) |
где Т - титр раствора йода, выраженный в граммах серы;
V - объем раствора йода, израсходованный на титрование, см3;
m - масса навески меди, г.
6.3.5.2 За результат анализа принимают среднеарифметическое значение двух параллельных определений при условии, что абсолютная разность между ними в условиях повторяемости не превышает значений (при доверительной вероятности Р = 0,95) предела повторяемости г, приведенных в таблице 1.
Если расхождение между результатами параллельных определений превышает значения предела повторяемости, выполняют процедуры, изложенные в стандарте [2] (подпункт 5.2.2.1).
6.3.6 Контроль точности результатов анализа
Контроль точности результатов анализа - по 3.14.
6.3.7 Оформление результатов анализа
Результаты анализа оформляют в соответствии с 3.15, значения погрешности результатов анализа А приведены в таблице 1.
6.4 Метод инфракрасной спектрометрии
6.4.1 Средства измерений, вспомогательные устройства, материалы, растворы
При выполнении измерений применяют следующие средства измерений, вспомогательные устройства:
- анализатор на серу, основанный на принципе инфракрасной спектрометрии с индукционной высокочастотной печью;
- печь шахтную, обеспечивающую температуру нагрева не менее 1200°С;
- тигли огнеупорные керамические, прокаленные при температуре от 900°С до 1200°С в течение не менее 4 ч;
- СО по ГОСТ 8.315 состава меди или сплавов на основе меди или на основе железа;
- пинцет медицинский по ГОСТ 21241.
При выполнении измерений применяют следующие материалы:
- кислород технический газообразный по ГОСТ 5583;
- магний хлорнокислый (ангидрон) фирмы LECO или по [17].
- плавни: вольфрам по [18], железо по [19] и другие вещества, обеспечивающие сжигание пробы и результаты контрольного опыта, указанные в 6.4.2.1;
- нити и волокна стеклянные однонаправленные по ГОСТ 10727;
- аскарит фирмы «LECO» или по [20].
6.4.2 Метод измерения
Метод основан на измерении светопоглощения газообразного оксида серы (IV) в инфракрасной области спектра после выделения его из навески металла сжиганием в индукционной высокочастотной печи в токе кислорода.
6.4.2.1 Измерение массовой доли серы (при массовой доле от 0,0003% до 0,050%) методом инфракрасной спектрометрии в присутствии плавня
Подготовка к выполнению измерений
Подготовку анализатора к работе и его градуировку проводят в соответствии с инструкцией по его эксплуатации. Для градуировки следует использовать стандартные образцы состава меди или сплавов на основе меди или на основе железа.
Выполнение измерений
Общие требования к методам измерений и требования безопасности при выполнении измерений в соответствии с разделами 3 и 4.
Массовую долю серы определяют параллельно из двух навесок.
В тигель помещают навеску анализируемой пробы массой от 0,2000 до 1,0000 г, добавляют плавень, масса которого должна быть одинаковой при проведении контрольного опыта, градуировки и анализа, и проводят анализ, как указано в прилагаемой к анализатору инструкции.
Непосредственно перед измерением навески анализируемой пробы проводят контрольный опыт. Для этого в тигель помещают навеску плавня такой массой, какую используют при анализе проб, и проводят анализ, как указано выше.
Контрольный опыт следует считать удовлетворительным, если показания массовой доли серы на цифровом дисплее не превышают погрешность метода анализа (таблица 2). Погрешностью метода анализа считают погрешность нижнего диапазона определяемых интервалов массовых долей серы.
Обработка результатов измерений
Результаты измерения массовой доли серы в процентах выводятся на дисплей или принтер автоматизированного анализатора.
За результат измерений принимают среднеарифметическое значение двух параллельных измерений при условии, что абсолютная разность между ними в условиях повторяемости не превышает значений (при доверительной вероятности Р = 0,95) предела повторяемости r, приведенных в таблице 2.
Если расхождение между результатами параллельных измерений превышает значение предела повторяемости, выполняют процедуры, изложенные в стандарте [2] (подпункт 5.2.2.1).
6.4.2.2 Измерение массовой доли серы (при массовой доле от 0,0002% до 0,0050%) методом инфракрасной спектрометрии без применения плавня
Подготовка к выполнению измерений
Подготовку анализатора к работе и его градуировку проводят в соответствии с инструкцией по его эксплуатации. Градуировку следует проводить по СО состава меди из трех параллельных измерений.
В случае градуировки анализатора предприятием-изготовителем повторная градуировка не требуется. В процессе применения данного анализатора проводят контроль стабильности градуировки в соответствии с инструкцией по эксплуатации.
Навески СО и анализируемого материала пробы должны быть одинаковыми.
Если найденное среднее значение массовой доли серы в СО отличается от аттестованного значения более чем на значение погрешности построения градуированной характеристики, градуировку повторяют, рассчитав линейный множитель для коррекции градуировки в соответствии с инструкцией по эксплуатации. При повторном превышении значения погрешности построения градуировочной характеристики проведение анализа прекращают до выяснения и устранения причин. Значение погрешности построения градуировочной характеристики устанавливают в лаборатории для конкретного экземпляра средства измерений.
Выполнение измерений
Общие требования к методам измерений и требования безопасности при выполнении измерений - в соответствии с разделами 3 и 4.
Массовую долю серы определяют из двух параллельных измерений.
В тигель помещают навеску анализируемой пробы массой (1,000 ±0,200) г, используя щипцы, ставят на подставку автопогрузочного устройства и далее проводят анализ, как указано в прилагаемой к анализатору инструкции.
Обработка результатов измерений
Результаты измерения массовой доли серы в процентах выводятся на дисплей компьютера.
За результат измерений принимают среднеарифметическое значение двух параллельных измерений при условии, что абсолютная разность между ними в условиях повторяемости не превышает значений (при доверительной вероятности Р = 0,95} предела повторяемости г, приведенных в таблице 3.
Если расхождение между результатами параллельных измерений превышает значение предела повторяемости, выполняют процедуры, изложенные в стандарте [2] (подпункт 5.2.2.1).
6.4.3 Контроль точности результатов измерений
Контроль точности результатов измерений - в соответствии с 3.14.
6.4.4 Оформление результатов измерений
Результаты измерений оформляют в соответствии с 3.15, значения погрешности результатов измерений ∆ приведены в таблицах 2 и 3.
7 Метод определения массовой доли фосфора
7.1 Область применения
В настоящем разделе установлено определение массовой доли фосфора в меди в диапазоне от 0,0003% до 0,06% фотометрическим методом.
Метод не распространяется на определение массовой доли фосфора в меди по ГОСТ 859 марок М00к и М00б.
7.2 Требования к погрешности анализа
Погрешность результатов анализа массовой доли фосфора, значения пределов повторяемости и воспроизводимости для доверительной вероятности Р = 0,95 должны соответствовать приведенным в таблице 4.
В процентах
|
Диапазон массовой доли фосфора |
Погрешность результатов анализа ±∆ |
Предел |
|
|
повторяемости r (n=2) |
воспроизводимости R |
||
|
От 0,0003 до 0,0010 включ. |
0,0002 |
0,0002 |
0,0003 |
|
Св. 0,0010 » 0,0030 » |
0,0003 |
0,0003 |
0,0004 |
|
» 0,0030 » 0,0100 » |
0,0006 |
0,0006 |
0,0008 |
|
» 0,010 » 0,030 » |
0,002 |
0,001 |
0,003 |
|
» 0,030 » 0,060 » |
0,004 |
0,002 |
0,005 |
7.3 Средства измерений, вспомогательные устройства, материалы, растворы
При выполнении анализа применяют следующие средства измерений, вспомогательные устройства:
- фотометр фотоэлектрический или спектрофотометр со всеми принадлежностями;
- весы лабораторные специального класса точности по ГОСТ 24104;
- пипетки не ниже 2-го класса точности по ГОСТ 29169 и ГОСТ 29227;
- колбы мерные 2-50-2, 2-100-2, 2-1000-2 по ГОСТ 1770;
- стаканы В-1-250 ТС по ГОСТ 25336;
- колбу Кн-1-100-14/23 по ГОСТ 25336;
- воронку Бюхнера по ГОСТ 9147;
- стекло часовое.
При выполнении анализа применяют следующие материалы, растворы:
- бумагу фильтровальную по ГОСТ 12026;
- фильтры обеззоленные по [21] или другие средней плотности;
- кислоту соляную по ГОСТ 3118;
- кислоту азотную по ГОСТ 4461 и разбавленную 2:1;
- смесь соляной и азотной кислот в соотношении 1:3, свежеприготовленную;
- аммоний ванадиевокислый мета по ГОСТ 9336, раствор массовой концентрации 2,5 г/дм3;
- аммоний молибденовокислый по ГОСТ 3765, перекристаллизованный, раствор массовой концентрации 100 г/дм3.
- водорода пероксид по ГОСТ 10929 и разбавленный 1:9;
- калий марганцовокислый по ГОСТ 20490, раствор массовой концентрации 0,2 моль/дм3 (1 н.);
- медь по ГОСТ 859;
- калий фосфорнокислый однозамещенный по ГОСТ 4198, высушенный при температуре от 80°С до 90°С в течение 1 ч;
- натрий фосфорнокислый двузамещенный по ГОСТ 11773, высушенный при температуре от 102°С до 105°С в течение 1 ч;
- растворы фосфора известной концентрации;
- аммиак водный по ГОСТ 3760;
- спирт этиловый ректификованный технический по ГОСТ 18300.
7.4 Метод анализа
Метод основан на образовании фосфорно-молибдено-ванадиевой гетерополикислоты в 1 М растворе азотной кислоты. Оптическую плотность раствора измеряют при длине волны от 400 до 413 нм или от 440 до 453 нм в зависимости от массовой доли фосфора.
7.5 Подготовка к выполнению анализа
7.5.1 При приготовлении раствора ванадиевокислого аммония массовой концентрации 2,5 г/дм3 навеску 2,5 г ванадиевокислого аммония растворяют в 650 см3 воды, добавляют 10 см3 азотной кислоты, доливают водой до 1000 см3 и перемешивают.
7.5.2 При приготовлении раствора молибденовокислого аммония массовой концентрации 100 г/дм3 вначале проводят перекристаллизацию соли следующим образом: навеску соли массой от 100 до 120 г растворяют в 400 см3 воды при температуре 80°С и дважды фильтруют горячий раствор через плотный обеззоленный фильтр «синяя лента». К полученному раствору добавляют 250 г этилового спирта, охлаждают и дают отстояться в течение 1 ч. Выпавшие кристаллы отфильтровывают на воронке Бюхнера. Полученные кристаллы молибденовокислого аммония растворяют и снова перекристаллизовывают, кристаллы отфильтровывают на воронке Бюхнера, промывают 2 - 3 раза этиловым спиртом объемом от 20 до 30 см3, после чего их высушивают на воздухе. Перед применением из перекристаллизованной соли готовят раствор следующим образом: навеску 100 г соли растворяют в воде объемом от 700 до 800 см3 и приливают от 25 до 30 см3 аммиака. Раствор перемешивают, затем его фильтруют через вату или бумажную массу, доливают водой до 1000 см3 и перемешивают. Используют свежеприготовленным.
7.5.3 Для построения градуировочных графиков готовят растворы фосфора известной концентрации. При приготовлении раствора А массовой концентрации фосфора 0,1 мг/см3 навеску 0,4580 г двузамещенного фосфорнокислого натрия или 0,4390 г однозамещенного фосфорнокислого калия растворяют в воде объемом от 50 до 70 см3, добавляют 2 см3 азотной кислоты, раствор переносят в мерную колбу вместимостью 1000 см3, доливают водой до метки и перемешивают.
При приготовлении раствора Б массовой концентрации фосфора 0,025 мг/см3 25 см3 раствора А помещают в мерную колбу вместимостью 100 см3, доливают водой до метки и перемешивают.
Растворы А и Б хранят в полиэтиленовой посуде. Раствор Б используют свежеприготовленным.
7.5.4 Построение градуировочных графиков
7.5.4.1 При массовой доле фосфора менее 0,001%
В мерные колбы вместимостью 50 см3 каждая помещают 0; 0,4; 1,0; 2,0; 3,0; 4,0 и 5,0 см3 раствора Б, что соответствует 0; 10; 25; 50; 75; 100 и 125 мкг фосфора, прибавляют от 3 до 4 см3 азотной кислоты, приливают 5 см3 ванадиевокислого аммония, 5 см3 раствора молибденовокислого аммония и доливают водой до метки. После прибавления каждого раствора содержимое колб хорошо перемешивают. Через 20 мин измеряют оптическую плотность раствора при длине волны от 400 до 413 нм в кювете толщиной поглощающего свет слоя 30 мм относительно раствора без добавления фосфора.
7.5.4.2 При массовой доле фосфора от 0,001% до 0,006% поступают так же, как в 7.5.4.1, однако количество раствора Б составляет 0; 1,0; 2,0; 4,0; 6,0; 8,0; 10,0 и 12,0 см3, что соответствует 0; 25; 50; 100; 150; 200; 250 и 300 мкг фосфора.
Оптическую плотность растворов измеряют при длине волны от 400 до 413 нм в кюветах толщиной поглощающего свет слоя 20 мм относительно раствора без добавления фосфора.
7.5.4.3 При массовой доле фосфора от 0,005% до 0,06%/
В мерные колбы вместимостью 100 см3 каждая помещают 0; 1,0; 2,5; 5,0; 7,5; 10,0; 12,0 и 13,0 см3 раствора А, что соответствует 0; 100; 250; 500; 750; 1000; 1200; 1300 мкг фосфора, прибавляют от 6 до 8 см3 азотной кислоты, 10 см3 ванадиевокислого аммония, 10 см3 раствора молибденовокислого аммония. После прибавления каждого раствора содержимое колб тщательно перемешивают. После этого раствор в мерной колбе немедленно доливают водой до метки и перемешивают. Через 20 мин измеряют оптическую плотность раствора при длине волны от 440 до 453 нм в кювете толщиной поглощающего свет слоя 30 мм относительно раствора без добавления фосфора.
7.5.4.4 При массовой доле фосфора от 0,01% до 0,06% в восьми стаканах вместимостью 250 см3 каждый взвешивают по 2,0000 г меди (с массовой долей фосфора менее 0,0005%), прибавляют 0; 1,0; 2,5; 5,0; 7,5; 10,0; 12,0 и 13,0 см3 раствора А, что соответствует 0; 100; 250; 500; 750; 1000; 1200; 1300 мкг фосфора. Растворы, при необходимости, выпаривают до объема от 1 до 2 см3. Затем в стаканы приливают по 30 см3 азотной кислоты, разбавленной 2:1. Раствор нагревают, не доводя до кипения, до полного растворения меди. После растворения пробы при слабом кипении удаляют оксиды азота, не снимая часовое стекло. Раствор охлаждают, прибавляют 1 см3 раствора марганцовокислого калия (до розовой окраски) и оставляют раствор на 5 мин. Затем нагревают до кипения, кипятят в течение 1 мин и охлаждают до температуры от 30°С до 40°С. Прибавляют 2 см3 пероксида водорода, разбавленного 1:9, кипятят в течение 30 с, затем прибавляют 10 см3 раствора ванадиевокислого аммония и продолжают кипятить в течение 1 мин.
Раствор охлаждают и переливают в мерную колбу вместимостью 100 см3.
Прибавляют 10 см3 раствора молибденовокислого аммония при непрерывном перемешивании. После этого раствор в мерной колбе немедленно доливают водой до метки и перемешивают. Через 20 мин измеряют оптическую плотность раствора при длине волны от 440 до 453 нм в кювете толщиной поглощающего свет слоя 30 мм.
Раствором сравнения при измерении оптической плотности служит раствор, содержащий 2 г меди (с массовой долей фосфора менее 0,0005%) и все реактивы.
7.5.4.5 При массовой доле фосфора от 0,001% до 0,06% с использованием смеси кислот в десять стаканов вместимостью 250 см3 каждый помещают 0; 0,8; 2,0 см3 раствора Б; 1,0; 2,5; 5,0; 7,5; 10,0; 12,0; 13,0 см3 раствора А, что соответствует 0; 20; 50; 100; 250; 500; 750; 1000; 1200; 1300 мкг фосфора, приливают от 18 до 20 см3 смеси соляной и азотной кислот в соотношении 1:3. Добавляют от 20 до 25 см3 воды и кипятят в течение 3 - 4 мин. Растворы охлаждают и помещают в мерные колбы вместимостью 50 см3.
К полученному раствору приливают при перемешивании 5 см3 раствора ванадиевокислого аммония и 5 см3 раствора молибденовокислого аммония, доливают водой до метки и перемешивают. Через 20 мин измеряют оптическую плотность раствора при длине волны от 440 до 453 нм в кювете толщиной поглощающего свет слоя 30 мм.
Раствором сравнения при измерении оптической плотности служит раствор, не содержащий фосфора.
По значениям оптических плотностей, найденных в 7.5.4.1 - 7.5.4.5, и соответствующим им значениям содержания фосфора строят градуировочные графики.
7.6 Выполнение анализа
7.6.1 Общие требования к методам анализа и требования безопасности при выполнении анализов - в соответствии с разделами 3 и 4.
7.6.2 Определение фосфора при массовой доле его от 0,0003% до 0,06%
Навеску меди массой от 2,0 до 5,0 г в зависимости от массовой доли фосфора (таблица 5) помещают в стакан вместимостью 250 см3, накрывают часовым стеклом и растворяют в 30 см3 азотной кислоты, разбавленной 2:1, при нагревании, не допуская кипения (при необходимости кислоту добавляют порциями по 10 см3).
|
Диапазон массовой доли фосфора. % |
Масса навески, г |
Объем азотной кислоты (2:1), см3 |
Объем анализируемого раствора, см3 |
Толщина поглощающего слоя, мм |
Длина волны, нм |
|
От 0,0003 до 0,001 |
5,0 |
30 (2:1) |
50 |
30 |
400 - 413 |
|
От 0,0005 до 0,006 |
5,0 |
30 (2:1) |
50 |
20 |
400 - 413 |
|
От 0,005 до 0,006 |
2,0 |
30 (2:1) |
100 |
30 |
440 - 453 |
Оксиды азота удаляют путем слабого кипячения раствора в замкнутом объеме (не снимая часового стекла). Раствор охлаждают, прибавляют 1 см3 раствора марганцовокислого калия (до розовой окраски) и оставляют раствор на 5 мин. Затем нагревают до кипения, кипятят в течение 1 мин и охлаждают до температуры от 30°С до 40°С. Прибавляют 2 см3 пероксида водорода, разбавленного 1:9, кипятят в течение 1 мин, прибавляют 5 см3 (или 10 см3 при разведении на 100 см3) раствора ванадиевокислого аммония и продолжают кипятить в течение 1 мин. Раствор охлаждают и переливают в зависимости от содержания фосфора (таблица 5) в мерную колбу вместимостью 50 см3 или 100 см3. При постоянном перемешивании по каплям прибавляют 5 см3 (10 см3) раствора молибденовокислого аммония. После этого раствор сразу доливают водой до метки и перемешивают.
Через 20 мин измеряют оптическую плотность раствора. Длина волны и толщина поглощающего свет слоя указаны в таблице 5. Раствором сравнения служит раствор, содержащий навеску меди и все реактивы, кроме молибденовокислого аммония.
Одновременно проводят два контрольных опыта, для чего в стакан вместимостью 250 см3 приливают 30 см3 азотной кислоты, разбавленной 2:1, накрывают часовым стеклом и проводят через ход анализа.
Раствором сравнения для контрольного опыта служит раствор, содержащий от 3 до 4 см3 (от 6 до 8 см3) азотной кислоты, 25 см3 воды и 5 см3 (10 см3) раствора ванадиевокислого аммония. Раствор переводят в мерную колбу вместимостью 50 см3 (100 см3) и доливают водой до метки.
Среднее значение оптической плотности растворов контрольных опытов вычитают из значения оптической плотности анализируемого раствора. Количество фосфора устанавливают по градуировочному графику, построенному, как указано в 7.5.4.1 - 7.5.4.3.
7.6.3 Определение фосфора при массовой доле его от 0,01% до 0,06% допускается проводить следующим образом.
Навеску меди массой 2,0 г помещают в стакан вместимостью 250 см3 и проводят определение по 7.6.2, измеряют оптическую плотность раствора при длине волны от 440 до 453 нм в кювете толщиной поглощающего свет слоя 30 мм. Раствором сравнения при измерении оптической плотности служит раствор, содержащий 2 г меди (с массовой долей фосфора менее 0,0005%), проведенный через ход анализа. Массу фосфора определяют по градуировочному графику, построенному в присутствии 2,0 г меди (с массовой долей фосфора менее 0,0005%), как указано в 7.5.4.4.
7.6.4 Определение фосфора при массовой доле его от 0,001% до 0,06% допускается проводить с использованием смеси кислот.
Навеску меди массой 2,0 г помещают в коническую колбу вместимостью 100 см3, приливают от 18 до 20 см3 смеси соляной и азотной кислот в соотношении 1:3, нагревают до растворения навески и далее нагрев продолжают до удаления окислов азота, не допуская кипения раствора. Затем добавляют от 20 до 25 см3 воды и кипятят от 3 до 4 мин. Раствор охлаждают и помещают в мерную колбу вместимостью 50 см3.
К полученному раствору приливают при перемешивании 5 см3 раствора ванадиевокислого аммония и 5 см3 раствора молибденовокислого аммония, доливают водой до метки и перемешивают. Через 20 мин измеряют оптическую плотность раствора при длине волны от 440 до 453 нм в кювете толщиной поглощающего свет слоя 30 мм.
Раствором сравнения при измерении оптической плотности служит раствор, не содержащий молибденовокислого аммония.
Одновременно через ход анализа проводят два контрольных опыта. Раствором сравнения служит раствор, не содержащий молибденовокислого аммония.
Среднее значение оптической плотности растворов контрольных опытов вычитают из значения оптической плотности анализируемого раствора.
Массу фосфора определяют по градуировочному графику, построенному, как указано в 7.5.4.5.
7.7 Обработка результатов анализа
7.7.1 Массовую долю фосфора X, %, вычисляют по формуле
|
|
(12) |
где m1 - масса фосфора, найденная по градуировочному графику, мкг;
т - масса навески меди, г.
7.7.2 За результат анализа принимают среднеарифметическое значение результатов двух параллельных определений при условии, что абсолютная разность между ними в условиях повторяемости не превышает значений (при доверительной вероятности Р = 0,95) предела повторяемости г, приведенных в таблице 4,
Если расхождение между результатами параллельных определений превышает значения предела повторяемости, выполняют процедуры, изложенные в стандарте [2] (подпункт 5.2.2,1).
7.8 Контроль точности результатов анализа
Контроль точности результатов анализа - по 3.14.
7.9 Оформление результатов анализа
Результаты анализа оформляют в соответствии с 3.15, значения погрешности результатов анализа ∆ приведены в таблице 4.
8 Методы определения массовой доли железа
8.1 Область применения
В настоящем разделе установлены фотометрический (при массовой доле от 0,0005% до 0,100%) и атомно-абсорбционный (при массовой доле от 0,0008% до 0,06%) методы определения массовой доли железа в меди.
8.2 Требования к погрешности анализа
Погрешность результатов анализа массовой доли железа, значения пределов повторяемости и воспроизводимости для доверительной вероятности Р - 0,95 должны соответствовать приведенным в таблице 6.
В процентах
|
Диапазон массовой доли железа |
Погрешность результатов анализа ±∆ |
Предел |
|
|
повторяемости r (n=2) |
воспроизводимости R |
||
|
От 0,0005 до 0,0010 включ. |
0,0002 |
0,0002 |
0,0003 |
|
Св. 0,0010 » 0,0030 » |
0,0003 |
0,0004 |
0,0006 |
|
» 0,003 » 0,010 » |
0,001 |
0,001 |
0,002 |
|
» 0,010 » 0,030 » |
0,002 |
0,002 |
0,005 |
|
» 0,030 » 0,100 » |
0,004 |
0,004 |
0,007 |
8.3 Фотометрический метод
8.3.1 Средства измерений, вспомогательные устройства, материалы, растворы
При выполнении анализа применяют следующие средства измерений, вспомогательные устройства:
- фотометр фотоэлектрический или спектрофотометр со всеми принадлежностями, обеспечивающими проведение измерений при длине волны 425 нм;
- центрифугу со всеми принадлежностями;
- весы лабораторные специального класса точности по ГОСТ 24104;
- пипетки не ниже 2-го класса точности по ГОСТ 29169 и ГОСТ 29227;
- колбы мерные 2-25-2, 2-50-2, 2-1000-2 по ГОСТ 1770;
- стаканы В-1-250 ТС, В-1-400 ТХС по ГОСТ 25336;
- стекло часовое.
При выполнении анализа применяют следующие материалы, растворы:
- кислоту соляную по ГОСТ 3118 и разбавленную 1:1;
- кислоту серную по ГОСТ 4204, разбавленную 1:4;
- воду бидистиллированную;
- кислоту азотную особой чистоты по ГОСТ 11125, разбавленную 1:1, или кислоту азотную по ГОСТ 4461 (прокипяченную для удаления окислов азота), разбавленную 1:1;
- аммиак водный по ГОСТ 3760, разбавленный 1:19;
- квасцы алюмокалиевые (алюминий-калий сернокислый) по ГОСТ 4329;
- алюминий первичный по ГОСТ 11069, марка А 999 или А 995;
- раствор алюминия;
- окись лантана;
- лантан азотнокислый шестиводный по [22] или лантан хлористый;
- раствор лантана массовой концентрации 1 мг/см3;
- кислоту сульфосалициловую по ГОСТ 4478, раствор массовой концентрации 100 г/дм3;
- аммоний хлористый по ГОСТ 3773, раствор массовой концентрации 200 г/дм3;
- железо карбонильное по [19] или другое, содержащее не менее 99,9% основного вещества;
- железа триоксид, предварительно высушенный при температуре 110°С;
- растворы железа известной концентрации.
8.3.2 Метод анализа
Метод основан на образовании желтого комплексного соединения железа с сульфосалициловой кислотой в аммиачном растворе после отделения железа от меди осаждением его с гидроксидом алюминия или лантана. Оптическую плотность раствора измеряют при длине волны 425 нм.
8.3.3 Подготовка к выполнению анализа
8.3.3.1 При приготовлении раствора алюминия навеску 1 г алюминия растворяют в объеме от 15 до 20 см3 соляной кислоты или навеску 20 г сернокислого алюминия-калия растворяют в воде с прибавлением 15 см3 соляной кислоты. Раствор доливают водой до 1000 см3 и перемешивают.
8.3.3.2 При приготовлении раствора лантана массовой концентрации 1 мг/см3 навеску 1,2 г окиси лантана растворяют в 15 см3 соляной кислоты, разбавленной 1:1, или навеску 2,7 г хлористого лантана или 3,1 г азотнокислого лантана растворяют в воде, прибавляют 10 см3 соляной кислоты, разбавленной 1:1. Раствор доливают водой до 1000 см3.
8.3.3.3 Для построения градуировочных графиков готовят растворы железа известной концентрации
При приготовлении раствора А массовой концентрации железа 0,1 мг/см3 навеску 0,1430 г триоксида железа или навеску 0,1000 г железа растворяют в 30 см3 соляной кислоты, разбавленной 1:1, при нагревании, При необходимости железо следует доокислить азотной кислотой, разбавленной 1:1. Раствор охлаждают и переносят в мерную колбу вместимостью 1000 см3, доливают водой до метки и перемешивают.
При приготовлении раствора Б массовой концентрации железа 0,02 мг/см3 20 см3 раствора А переносят пипеткой в мерную колбу вместимостью 100 см3, приливают 2 см3 соляной кислоты, разбавленной 1:1, доливают до метки водой и перемешивают.
8.3.3.4 Построение градуировочного графика
В стаканы помещают 0; 0,2; 0,5; 1,0; 2,0; 3,0; 4,0 и 5,0 см3 раствора Б, что соответствует 0; 4; 10; 20; 40; 80; 80 и 100 мкг железа, прибавляют 5 см3 азотной кислоты, 25 см3 воды, 5 см3 раствора алюминия или лантана. Отделение железа, растворение гидроксидов соляной кислотой и измерение оптической плотности растворов выполняют, как указано в 8.3.4.2.
По полученным значениям оптической плотности и соответствующим им содержаниям железа строят градуировочный график.
8.3.4 Выполнение анализа
8.3.4.1 Общие требования к методам анализа и требования безопасности при выполнении анализов - в соответствии с разделами 3 и 4.
8.3.4.2 Определение железа при массовой доле его от 0,0005% до 0,01%
Навеску меди массой 1,0000 г помещают в стакан вместимостью 100 см3 и растворяют в 5 см3 азотной кислоты. Окислы азота удаляют при осторожном кипячении в стакане, прикрытом часовым стеклом. Раствор разбавляют 25 см3 воды, прибавляют 5 см3 раствора алюминия или лантана, затем при постоянном перемешивании раствор аммиака в таком количестве, чтобы вся медь перешла в комплексное соединение (синий раствор). Раствор с осадком нагревают до температуры от 70°С до 80°С и выдерживают при этой температуре в течение 20 мин. После охлаждения гидроксиды отделяют фильтрованием или центрифугированием.
Для центрифугирования содержимое стакана переливают в пробирку центрифуги и центрифугируют в течение 2 мин. Затем раствор над осадком сливают (сифонируют), а осадок в пробирке два раза промывают по 10 см3 раствора аммиака, разбавленного 1:19, каждый раз сливая промывной раствор. К осадку в пробирке прибавляют 2 см3 горячей соляной кислоты, разбавленной 1:1, и после растворения осадка прибавляют 10 см3 воды. Затем при перемешивании прибавляют по каплям раствор аммиака до осаждения гидроксидов. Через 10 мин содержимое пробирки центрифугируют, и раствор над осадком сливают. Осадок в пробирке два раза промывают по 10 см3 раствора аммиака, разбавленного 1:19, растворяют в 5 см3 соляной кислоты, разбавленной 1:1, и раствор переносят в стакан, в котором проводилось осаждение.
Содержимое стакана после осаждения гидроксидов фильтруют на фильтр «белая лента». Осадок на фильтре промывают 5 - 6 раз горячим раствором аммиака, разбавленным 1:19. Затем осадок смывают с фильтра струей горячей воды в стакан, в котором проводилось осаждение, прибавляют 5 см3 соляной кислоты и содержимое стакана нагревают до растворения осадка (раствор должен быть прозрачным). Раствор в стакане охлаждают, прибавляют 25 см3 воды и переосаждают гидроксиды раствором аммиака.
Осадок гидроксидов фильтруют на тот же фильтр и промывают на фильтре 5 - 6 раз горячим раствором аммиака, разбавленным 1:19. Затем осадок с фильтра смывают струей горячей воды в стакан, в котором проводили осаждение. Осадок гидроксидов на фильтре растворяют в объеме от 5 до 10 см3 соляной кислоты и собирают раствор в стакан, в котором осаждали гидроксиды. Фильтр промывают 2 - 3 раза малыми порциями горячей воды, присоединяя промывные воды к основному раствору в стакане.
Раствор выпаривают до объема от 2 до 3 см3 и, после охлаждения, переливают в мерную колбу вместимостью 50 см3. Стакан обмывают раствором хлористого аммония 2 раза по 5 см3. К раствору в мерной колбе прибавляют 2,5 см3 раствора сульфосалициловой кислоты, перемешивают, прибавляют 5 см3 раствора аммиака и доливают водой до метки. Оптическую плотность раствора измеряют не позже, чем через 30 мин при длине волны 425 нм в кювете толщиной поглощающего свет слоя 50 мм. Раствором сравнения при измерении оптической плотности служит вода.
Одновременно проводят два контрольных опыта со всеми применяемыми реактивами.
Среднее значение оптической плотности растворов контрольных опытов вычитают из значения оптической плотности анализируемого раствора.
Массу железа в растворе устанавливают по градуировочному графику, построенному, как указано в 8.3.3.4.
8.3.4.3 Определение железа при массовой доле его от 0,01% до 0,1%
Растворение и отделение железа выполняют таким же образом, как описано в 8.3.4.2. К солянокислому раствору, полученному после растворения гидроксидов, прибавляют 20 см3 соляной кислоты, разбавленной 1:1, раствор переносят в мерную колбу вместимостью 50 см3, доливают водой до метки и перемешивают; 5 см3 этого раствора переносят пипеткой в мерную колбу вместимостью 25 см3, прибавляют 10 см3 раствора хлористого аммония, 2,5 см3 раствора сульфосалициловой кислоты, перемешивают, прибавляют 5 см3 аммиака, доливают водой до метки и перемешивают. Далее поступают так же, как указано в 8.3.4.2.
8.4 Атомно-абсорбционный метод
8.4.1 Средства измерений, вспомогательные устройства, материалы, растворы
При выполнении анализа применяют следующие средства измерений, вспомогательные устройства:
- спектрофотометр атомно-абсорбционный с источником излучения на железо;
- компрессор воздушный;
- весы лабораторные специального класса точности по ГОСТ 24104;
- пипетки не ниже 2-го класса точности по ГОСТ 29169 и ГОСТ 29227;
- колбы мерные 2-25-2, 2-100-2, 2-1000-2 по ГОСТ 1770;
- колбы Кн-2-100-14/23 ТХС, Кн-2-250- 19/26 ТХС по ГОСТ 25336;
- стаканы В-1- 250 ТХС по ГОСТ 25336.
При выполнении анализа применяют следующие материалы, растворы:
- ацетилен по ГОСТ 5457;
- воду бидистиллированную;
- кислоту азотную особой чистоты по ГОСТ 11125, разбавленную 1:1, или кислоту азотную по ГОСТ 4461 (прокипяченную для удаления окислов азота), разбавленную 1:1;
- медь, СО для спектрального анализа, содержащий 6,8•10-4 % железа, или электролитную медь с установленной массовой долей железа;
- железо карбонильное по [19] или другое, содержащее не менее 99,9% основного вещества;
- растворы железа известной концентрации.
8.4.2 Метод анализа
Атомно-абсорбционный метод основан на растворении пробы в азотной кислоте и последующем измерении при длине волны 248,3 нм поглощения резонансного излучения атомами железа при введении солянокислого или азотнокислого раствора в пламя ацетилен-воздух.
При массовой доле железа до 0,002% его отделяют от основного количества меди соосаждением на гидроксиде лантана.
8.4.3 Подготовка к выполнению анализа
8.4.3.1 Приготовление растворов железа известной концентрации
При приготовлении раствора А массовой концентрации железа 0,1 мг/см3 навеску 0,1000 г железа растворяют в 10 см3 азотной кислоты, разбавленной 1:1, раствор переносят в мерную колбу вместимостью 1000 см3, доливают водой до метки и перемешивают.
При приготовлении раствора Б массовой концентрации железа 0,01 мг/см3 10 см3 раствора А помещают в мерную колбу вместимостью 100 см3, доливают до метки водой и перемешивают.
8.4.3.2 Построение градуировочного графика
Для построения градуировочного графика в мерные колбы вместимостью 100 см3 каждая помещают 1,0 г СО меди, приливают 10 см3 азотной кислоты, разбавленной 1:1. После растворения меди в мерные колбы помещают 0; 0,5; 1; 2; 5; 10; 20 и 50 см3 раствора Б, доливают водой до метки и перемешивают.
Полученные растворы содержат С, С+5, С+10, С+20, С+50, С+100, С+200, С+500 мкг железа, где С - содержание железа (мкг) в СО меди, взятом в качестве фонового для построения градуировочного графика.
Измеряют абсорбцию приготовленных растворов, как указано в 8.4.4.2.
По полученным значениям оптических плотностей растворов и соответствующим им концентрациям железа строят градуировочный график.
При построении графика значение сигнала фонового раствора необходимо вычесть из значения сигнала каждого раствора известной концентрации и провести график из начала координат.
8.4.4 Выполнение анализа
8.4.4.1 Общие требования к методам анализа и требования безопасности при выполнении анализов - в соответствии с разделами 3 и 4.
8.4.4.2 При массовой доле железа от 0,002% до 0,06% навеску меди массой 1,0000 г помещают в коническую колбу вместимостью 100 см3 и растворяют в 10 см3 азотной кислоты, разбавленной 1:1. Раствор переводят в мерную колбу вместимостью 100 см3, доливают до метки водой и перемешивают. Полученный раствор меди распыляют в пламя и измеряют абсорбцию при длине волны 248,3 нм.
Одновременно проводят контрольный опыт со всеми применяемыми реактивами. Значение оптической плотности раствора контрольного опыта вычитают из значения оптической плотности анализируемого раствора.
Массу железа в растворе определяют по градуировочному графику.
Допускается для определения массовой доли железа использовать метод добавок.
8.4.4.3 При массовой доле железа от 0,0008% до 0,002% навеску меди массой 1,0000 г помещают в стакан или коническую колбу вместимостью 250 см3 и растворяют в объеме от 10 до 15 см3 азотной кислоты, разбавленной 1:1, и далее анализ проводят по 8.3.4.2.
Раствор после отделения меди выпаривают до объема от 6 до 8 см3, охлаждают, помещают в мерную колбу вместимостью 25 см3, разбавляют водой до метки и перемешивают.
Измеряют абсорбцию при длине волны 248,3 нм, вводя анализируемый раствор в пламя ацетилен-воздух.
Массу железа в растворе определяют по градуировочному графику.
Допускается определение в анализируемом растворе цинка (от 0,0005% до 0,006%), олова (от 0,005% до 0,06%), никеля (от 0,1% до 0,5%) (при анализе без отделения меди аммиаком).
8.5 Обработка результатов анализа
8.5.1 Массовую долю железа X, %, при использовании фотометрического метода определения железа вычисляют по формулам:
- при массовой доле железа от 0,0005% до 0,01%
|
|
(13) |
- при массовой доле железа от 0,01 % до 0,1 %
|
|
(14) |
где т1 - масса железа, найденная по градуировочному графику, мкг;
V - объем анализируемого раствора, см3;
V1 - объем аликвотной части анализируемого раствора, см3;
т - масса навески меди, г.
8.5.2 Массовую долю железа Х, %, при использовании атомно-абсорбционного метода определения массовой доли железа вычисляют по формуле
|
|
(15) |
где m1 - масса железа, найденная по градуировочному графику, мкг;
т - масса навески меди, г.
8.5.3 За результат анализа принимают среднеарифметическое значение двух параллельных определений при условии, что абсолютная разность между ними в условиях повторяемости не превышает значений (при доверительной вероятности Р = 0,95) предела повторяемости г, приведенных в таблице 6.
Если расхождение между результатами параллельных определений превышает значение предела повторяемости, выполняют процедуры, изложенные в стандарте [2] (подпункт 5.2.2.1).
8.6 Контроль точности результатов анализа
Контроль точности результатов анализа - по 3.14.
8.7 Оформление результатов анализа
Результаты анализа оформляют в соответствии с 3.15, значения погрешности результатов анализа ∆ приведены в таблице 6.
9 Метод определения массовой доли цинка
9.1 Область применения
Настоящий раздел устанавливает определение массовой доли цинка в меди в диапазоне от 0,0005% до 0,006% атомно-абсорбционным методом.
Метод не распространяется на определение массовой доли цинка в меди по ГОСТ 859 марок М00к и М00б.
9.2 Требования к погрешности анализа
Погрешность результатов анализа массовой доли цинка, значения пределов повторяемости и воспроизводимости для доверительной вероятности Р = 0,95 должны соответствовать приведенным в таблице 7.
В процентах
|
Диапазон массовой доли цинка |
Погрешность результатов анализа ±∆ |
Предел |
|
|
повторяемости r (n=2) |
воспроизводимости R |
||
|
От 0,0005 до 0,0010 включ. |
0,0002 |
0,0002 |
0,0003 |
|
Св. 0,0010 » 0,0030 » |
0,0003 |
0,0004 |
0,0005 |
|
» 0,0030 » 0,0060 » |
0,0005 |
0,0006 |
0,0007 |
9.3 Средства измерений, вспомогательные устройства, материалы, растворы
При выполнении анализа применяют следующие средства измерений, вспомогательные устройства:
- спектрофотометр атомно-абсорбционный с источником излучения на цинк;
- компрессор воздушный;
- весы лабораторные специального класса точности по ГОСТ 24104;
- пипетки не ниже 2-го класса точности по ГОСТ 29169 и ГОСТ 29227;
- колбы мерные 2-100-2, 2-1000-2 по ГОСТ 1770;
- стаканы В-1-250 ТХС по ГОСТ 25336;
- колбы Кн-2-250-19/26 ТХС по ГОСТ 25336.
При выполнении анализа применяют следующие материалы, растворы:
- ацетилен по ГОСТ 5457;
- воду бидистиллированную;
- кислоту азотную особой чистоты по ГОСТ 11125, разбавленную 1:1, или кислоту азотную по ГОСТ 4461 (прокипяченную до удаления окислов азота);
- медь по ГОСТ 859;
- цинк по ГОСТ 3640;
- растворы цинка известной концентрации.
9.4 Метод анализа
Метод основан на растворении пробы в азотной кислоте и измерении при длине волны 213 нм поглощения резонансного излучения атомами цинка, образующимися в результате атомизации при введении раствора пробы в пламя ацетилен-воздух.
9.5 Подготовка к выполнению анализа
9.5.1 Для построения градуировочных графиков готовят растворы цинка известной концентрации.
При приготовлении раствора А массовой концентрации цинка 0,1 мг/см3 навеску 0,100 г цинка растворяют в 10 см3 азотной кислоты, разбавленной 1:1, раствор переводят в мерную колбу вместимостью 1000 см3, доливают водой до метки и перемешивают.
При приготовлении раствора Б массовой концентрации цинка 0,01 мг/см3 10 см3 раствора А помещают в мерную колбу вместимостью 100 см3, доливают до метки водой и перемешивают.
9.5.2 Построение градуировочного графика
Для построения градуировочного графика в мерные колбы вместимостью 100 см3 каждая помещают 0; 0,5; 1,0; 2,0; 4,0; и 6,0 см3 раствора Б, что соответствует 0; 5; 10; 20; 40; 60 мкг цинка. Во все колбы прибавляют по 2 см3 раствора азотной кислоты, доливают водой до метки и перемешивают.
Измеряют абсорбцию приготовленных растворов, как указано в 9.6.2.
По полученным значениям оптических плотностей растворов и соответствующим им значениям содержания цинка строят градуировочный график. При построении графика значения сигнала фонового раствора вычитают из значения сигнала раствора известной концентрации и проводят график из начала координат.
9.6 Выполнение анализа
9.6.1 Общие требования к методам анализа и требования безопасности при выполнении анализа - в соответствии с разделами 3 и 4.
9.6.2 Навеску меди массой 1,0000 г помещают в стакан вместимостью 100 см3 и растворяют при нагревании в 10 см3 азотной кислоты, разбавленной 1:1, до удаления окислов азота. Раствор охлаждают, переводят в мерную колбу вместимостью 100 см3, доливают до метки водой и перемешивают. Полученный раствор меди распыляют в пламя атомно-абсорбционного спектрофотометра и измеряют абсорбцию при длине волны 213,8 нм.
Допускается определение в анализируемом растворе никеля (от 0,1% до 0,5%), свинца (от 0,005% до 0,06%), кобальта (от 0,005% до 0,2%), железа (от 0,01% до 0,08%).
Одновременно проводят контрольный опыт со всеми применяемыми реактивами. Значение оптической плотности раствора контрольного опыта вычитают из значения оптической плотности анализируемого раствора.
Массу цинка в растворе определяют по градуировочному графику.
Допускается для определения массовой доли цинка использовать метод добавок.
9.7 Обработка результатов анализа
9.7.1 Массовую долю цинка X, %, вычисляют по формуле
|
|
(16) |
где т1 - масса цинка, найденная по градуировочному графику, мкг;
т - масса навески меди, г.
9.7.2 За результат анализа принимают среднеарифметическое значение двух параллельных определений при условии, что абсолютная разность между ними в условиях повторяемости не превышает значений (при доверительной вероятности Р = 0,95) предела повторяемости г, приведенных в таблице 7.
Если расхождение между результатами параллельных определений превышает значение предела повторяемости, выполняют процедуры, изложенные в стандарте [2] (подпункт 5.2.2.1).
9.8 Контроль точности результатов анализа
Контроль точности результатов анализа - по 3.14.
9.9 Оформление результатов анализа
Результаты анализа оформляют в соответствии с 3.15, значения погрешности результатов анализа А приведены в таблице 7.
10 Методы определения массовой доли никеля
10.1 Область применения
В настоящем разделе установлены фотометрический (при массовой доле от 0,0005% до 0,5%) и атомно-абсорбционный (при массовой доле от 0,0005% до 0,4%) методы определения массовой доли никеля в меди.
10.2 Требования к погрешности анализа
Погрешность результатов анализа массовой доли никеля, значения пределов повторяемости и воспроизводимости для доверительной вероятности Р = 0,95 должны соответствовать приведенным в таблице 8.
В процентах
|
Диапазон массовой доли никеля |
Погрешность результатов анализа ±∆ |
Предел |
|
|
повторяемости r (n=2) |
воспроизводимости R |
||
|
От 0,0005 до 0,0010 включ. |
0,0002 |
0,0002 |
0,0003 |
|
Св. 0,0010 » 0,0030 » |
0,0004 |
0,0004 |
0,0005 |
|
» 0,003 » 0,010 » |
0,001 |
0,001 |
0,002 |
|
» 0,010 » 0,030 » |
0,002 |
0,002 |
0,003 |
|
» 0,030 » 0,100 » |
0,004 |
0,004 |
0,006 |
|
» 0,10 » 0,30 » |
0,01 |
0,01 |
0,02 |
|
» 0,30 » 0,50 » |
0,04 |
0,04 |
0,06 |
10.3 Фотометрический метод
10.3.1 Средства измерений, вспомогательные устройства, материалы, растворы
При выполнении анализа применяют следующие средства измерений, вспомогательные устройства:
- фотометр фотоэлектрический или спектрофотометр, обеспечивающий проведение измерений в области длин волн от 400 до 450 нм;
- рН-метр со всеми принадлежностями;
- весы лабораторные специального класса точности по ГОСТ 24104;
- пипетки не ниже 2-го класса точности по ГОСТ 29169 и ГОСТ 29227;
- колбы мерные 2-25-2, 2-50-2, 2-100-1, 2-1000-2 по ГОСТ 1770;
- стаканы В-1- 50 ТХС, В-1-100 ТХС, В-1-250 ТС, В-1-400 ТХС по ГОСТ 25336;
- воронку ВД-1-100 ХС по ГОСТ 25336.
При выполнении анализа применяют следующие материалы, растворы:
- кислоту соляную по ГОСТ 3118, раствор молярной концентрации 0,5 моль/дм3 и разбавленную 1:3;
- кислоту серную по ГОСТ 4204, разбавленную 1:1;
- кислоту азотную по ГОСТ 4461, разбавленную 1:1 и 3:2;
- аммиак водный по ГОСТ 3760, разбавленный 1:1;
- бром по ГОСТ 4109, насыщенный водный раствор (бромная вода);
- диметилглиоксим по ГОСТ 5828, спиртовой свежеприготовленный раствор массовой концентрации 5 г/дм3 и раствор массовой концентрации 10 г/дм3 в растворе гидроксида натрия массовой концентрации 80 г/дм3;
- натрия гидроксид (натрия гидроокись) по ГОСТ 4328, растворы массовой концентрации 80,100 и 400 г/дм3;
- натрий углекислый безводный по ГОСТ 83, раствор массовой концентрации 500 г/дм3 и раствор массовой концентрации 10 г/дм3 в растворе гидроксида натрия массовой концентрации 400 г/дм3;
- натрий серноватистокислый (тиосульфат натрия) 5-водный по ГОСТ 27068, раствор массовой концентрации 500 г/дм3;
- натрий уксуснокислый по ГОСТ 199;
- ацетатный буферный раствор с рН 6,5 ±0,3;
- тартратный буферный раствор с рН 6,5 ±0,3;
- спирт этиловый ректификованный по ГОСТ 18300;
- хлороформ по ГОСТ 20015;
- йод по ГОСТ 4159, спиртовой раствор массовой концентрации 10 г/дм3;
- никель по ГОСТ 849;
- никель (II) сернокислый 7-водный по ГОСТ 4465;
- растворы никеля известной концентрации;
- водорода пероксид по ГОСТ 10929;
- медь по ГОСТ 859 (с массовой долей никеля не более 0,0005%);
- растворы меди;
- калий-натрий виннокислый (сегнетова соль) по ГОСТ 5845, раствор массовой концентрации 200 г/дм3;
- аммоний надсернокислый по ГОСТ 20478, раствор массовой концентрации 30 г/дм3;
- смесь соляной и азотной кислот в отношении 3:1, свежеприготовленную;
- кислоту лимонную по ГОСТ 3652, раствор массовой концентрации 100 г/дм3;
- кислоту уксусную по ГОСТ 61;
- кислоту винную по ГОСТ 5817;
- бумагу фильтровальную по ГОСТ 12026.
10.3.2 Метод анализа
Метод основан на образовании окрашенного соединения никеля с диметилглиоксимом в среде аммиака или гидроксида натрия после отделения никеля в виде диметилглиоксимата экстракцией хлороформом и реэкстракции никеля соляной кислотой. Оптическую плотность раствора измеряют при длине волны от 434 до 450 нм. Медь связывают тиосульфатом натрия в бесцветный комплекс при рН от 8,2 до 6,8.
10.3.3 Подготовка к выполнению анализа
10.3.3.1 При приготовлении ацетатного буферного раствора навеску 300 г уксуснокислого натрия растворяют в 800 см3 воды и устанавливают рН раствора (6,5 ±0,3) прибавлением уксусной кислоты. Раствор переводят в мерную колбу вместимостью 1000 см3, доливают водой до метки и перемешивают.
10.3.3.2 При приготовлении тартратного буферного раствора навеску 150 г винной кислоты растворяют в 800 см3 воды и устанавливают рН раствора (8,5 ±0,3) прибавлением раствора гидроксида натрия массовой концентрации 400 г/дм3. Раствор переводят в мерную колбу вместимостью 1000 см3, доливают до метки водой и перемешивают.
10.3.3.3 Для построения градуировочных графиков готовят растворы никеля известной концентрации.
При приготовлении раствора А массовой концентрации никеля 1 мг/см3 навеску 4,7850 г сернокислого никеля помещают в стакан вместимостью 250 см3, прибавляют 50 см3 воды, 10 см3 серной кислоты, переводят в мерную колбу вместимостью 1000 см3, доливают водой до метки и перемешивают.
При применении металлического никеля навеску 1,0000 г никеля при нагревании растворяют в 50 см3 соляной кислоты и 20 см3 пероксида водорода, раствор охлаждают, приливают 10 см3 серной кислоты, разбавленной 1:1, и раствор выпаривают до появления паров серной кислоты. Остаток охлаждают, приливают 100 см3 воды, переводят раствор в мерную колбу вместимостью 1000 см3, полученный раствор доливают до метки водой и перемешивают.
Такой же раствор может быть приготовлен следующим образом: навеску никеля массой 1,0000 г растворяют при нагревании в объеме от 20 до 25 см3 азотной кислоты, разбавленной 3:2, и выпаривают раствор до объема от 3 до 5 см3. Затем добавляют 10 см3 серной кислоты, разбавленной 1:1, и выпаривают до выделения паров серной кислоты. После охлаждения приливают от 100 до 150 см3 воды, растворяют соли, переносят раствор в мерную колбу вместимостью 1000 см3, доводят водой до метки и перемешивают.
При приготовлении раствора Б массовой концентрации никеля 0,1 мг/см3100 см3 раствора А помещают в мерную колбу вместимостью 1000 см3, приливают 1 см3 серной кислоты, разбавленной 1:1, доливают водой до метки и перемешивают.
При приготовлении раствора В массовой концентрации никеля 0,01 мг/см3 25 см3 раствора Б помещают в мерную колбу вместимостью 250 см3, разбавляют водой до метки и перемешивают.
Растворы Б и В применяют свежеприготовленными.
10.3.3.4 Для построения градуировочных графиков готовят растворы меди известной концентрации.
При приготовлении раствора А массовой концентрации меди 0,1 мг/см3 навеску 25,0 г меди растворяют в 200 см3 азотной кислоты, разбавленной 1:1, раствор нагревают до удаления окислов азота, охлаждают и переводят в мерную колбу вместимостью 250 см3, доливают водой до метки и перемешивают.
При приготовлении раствора Б массовой концентрации меди 0,01 мг/см3 25 см3 раствора А помещают в мерную колбу вместимостью 250 см3, разбавляют водой до метки и перемешивают. Раствор устойчив в течение 8 ч.
10.3.3.5 Построение градуировочного графика при массовой доле никеля от 0,0005% до 0,005%
В стаканы вместимостью 100 см3 каждый отбирают по 10 см3 раствора меди А и 0; 0,5; 1,0; 2,0; 3,0; 4,0 и 5,0 см3 раствора никеля В, что соответствует 0; 5; 10; 20; 30; 40 и 50 мкг никеля. Контрольным опытом служит раствор меди без добавления раствора никеля. Растворы выпаривают примерно до объема 3 см3. Остаток растворяют в 10 см3 воды и далее поступают так, как указано в 10.3.4.2 и 10.3.4.3.
10.3.3.6 Построение градуировочного графика при массовой доле никеля от 0,005% до 0,05%
В стаканы вместимостью 100 см3 каждый отбирают по 10 см3 раствора меди А и 0; 0,5; 1,0; 2,0; 3,0; 4,0 и 5,0 см3 раствора никеля Б, что соответствует 0; 50; 100; 200; 300; 400 и 500 мкг никеля. Контрольным опытом служит раствор меди без добавления стандартного раствора никеля. Растворы выпаривают примерно до объема 3 см3. Остаток растворяют в 10 см3 воды, переводят раствор в делительную воронку вместимостью 100 см3 и далее проводят анализ так, как указано в 10.3.4.4.
10.3.3.7 Построение градуировочного графика при массовой доле никеля от 0,05% до 0,5%
В стаканы вместимостью 100 см3 каждый отбирают по 10 см3 раствора меди Б и 0; 0,5; 1,0; 2,0; 3,0; 4,0 и 5,0 см3 раствора никеля Б, что соответствует 0; 50; 100; 200; 300; 400 и 500 мкг никеля. Контрольным опытом служит раствор меди без добавления раствора никеля. Растворы выпаривают примерно до объема 3 см3. Остаток растворяют в 10 см3 воды, переводят раствор в делительную воронку вместимостью 100 см3 и далее проводят анализ так, как указано в 10.3.4.5.
По значениям оптических плотностей растворов, найденным в 10.3.3.5; 10.3.3.6 и 10.3.3.7, и соответствующим им содержаниям никеля строят градуировочные графики.
10.3.3.8 Построение градуировочного графика для определения никеля в электролите после выделения меди в соответствии с разделом 5.
В мерные колбы вместимостью 50 см3 каждая помещают соответственно 0; 0,5; 1,0; 2,0; 4,0; 5,0 и 10,0 см3 раствора Б или В и далее продолжают анализ, как описано в 10.3.4.5. По полученным значениям оптических плотностей и соответствующим им концентрациям растворов строят график.
10.3.4 Выполнение анализа
10.3.4.1 Общие требования к методам анализа и требования безопасности при выполнении анализов - в соответствии с разделами 3 и 4.
10.3.4.2 Определение никеля при массовой доле его от 0,0005% до 0,005%
Навеску меди массой 1,0000 г помещают в стакан вместимостью 250 см3 и растворяют в 10 см3 азотной кислоты, разбавленной 1:1. После прекращения бурной реакции раствор осторожно кипятят до удаления окислов азота в течение 7 - 10 мин, выпаривают примерно до объема 3 см3, к раствору прибавляют 10 см3 воды и нагревают до кипения. Раствор охлаждают, прибавляют раствор аммиака, разбавленный 1:1, до появления осадка гидроксидов, затем по каплям соляную кислоту, разбавленную 1:3, до растворения осадка. К раствору приливают 3 см3 тартратного буферного раствора, 3 см3 ацетатного буферного раствора и 25 см3 раствора серноватистокислого натрия. Величину рН раствора (6,5 ±0,3) контролируют при помощи рН-метра. Раствор переводят в делительную воронку вместимостью 100 см3, приливают 2 см3 спиртового раствора диметилглиоксима, перемешивают, и раствор выдерживают в течение от 1 до 2 мин.
К содержимому воронки прибавляют 5 см3 (для анализа по 10.3.4.2) или 10 см3 (для анализа по 10.3.4.3) хлороформа и экстрагируют соединение никеля в течение 1 мин. Хлороформный экстракт отделяют и помещают в другую делительную воронку. К водному раствору в воронке прибавляют еще 5 см3 (для анализа по 10.3.4.2) или 10 см3 (для анализа по 10.3.4.3) хлороформа и повторяют экстракцию. Сливают экстракты в стакан, в котором проводили разложение пробы, аммиачные растворы отбрасывают.
К объединенным хлороформным экстрактам прибавляют 5 см3 (для анализа по 10.3.4.2) или 10 см3 (для анализа по 10.3.4.3) 0,5 моль/дм3 раствора соляной кислоты и реэкстрагируют никель в течение 1 мин, хлороформ сливают в другую делительную воронку и повторяют еще два раза. Реэкстракты помещают в стакан вместимостью 100 см3.
При работе в среде аммиака реэкстракты помещают в стакан вместимостью 100 см3, нагревают до кипения и упаривают до объема от 7 до 10 см3, охлаждают, помещают в мерную колбу вместимостью 25 или 50 см3 и последовательно прибавляют 2 см3 спиртового раствора диметилглиоксима, 5 см3 раствора надсернокислого аммония и 5 или 10 см3 аммиака, разбавленного 1:1. Разбавляют водой до метки и перемешивают. Через 10 мин измеряют оптическую плотность раствора при длине волны 445 нм в кюветах толщиной поглощающего свет слоя 50 мм. Раствором сравнения служит вода.
При работе в среде гидроксида натрия реэкстракты помещают в стакан вместимостью 100 см3 и последовательно прибавляют 1 см3 раствора лимонной кислоты, 2 см3 раствора надсернокислого аммония, 10 см3 раствора гидроксида натрия (80 г/дм3) и 1 см3 раствора диметилглиоксима в растворе гидроксида натрия. Раствор нагревают до 60°С и оставляют при этой температуре на 5 мин. Затем охлаждают, помещают в мерную колбу вместимостью 50 см3, разбавляют водой до метки и перемешивают. Оптическую плотность измеряют, как описано выше.
Массу никеля определяют по градуировочному графику, построенному, как указано в 10.3.3.5.
Одновременно проводят два контрольных опыта, выполняя те же операции и приливая те же реактивы, что и при анализе пробы. Среднее значение оптической плотности раствора контрольного опыта вычитают из значения оптической плотности анализируемого раствора.
10.3.4.3 Для меди, содержащей соединения никеля, не растворимые в азотной кислоте, разбавленной 1:1, вскрытие навески осуществляют следующим образом: навеску меди массой 1,0000 г помещают в стакан вместимостью 250 см3, приливают 20 см3 смеси соляной и азотной кислот и выпаривают при нагревании до влажных солей. Затем добавляют 10 см3 соляной кислоты и выпаривают досуха. Эту операцию повторяют еще два раза. После охлаждения сухой остаток смачивают 3 см3 соляной кислоты, приливают 10 см3 воды и нагревают до растворения солей. Раствор охлаждают, прибавляют аммиак, разбавленный 1:1, до появления осадка гидроксидов и затем по каплям раствор соляной кислоты 0,5 моль/дм3 до растворения осадка. К полученному раствору приливают 3 см3 тартратного буферного раствора и от 3 до 5 см3 ацетатного буферного раствора до растворения осадка. Добавляют 35 см3 раствора тиосульфата натрия и затем еще по каплям до обесцвечивания раствора (до полного восстановления меди). Далее продолжают анализ по 10.3.4.2.
10.3.4.4 Определение никеля при массовой доле его от 0,005% до 0,05%
Навеску меди массой 1,0000 г помещают в стакан вместимостью 250 см3 и поступают далее, как указано в 10.3.4.2.
Для экстракции соединения никеля используют дважды по 10 см3 хлороформа и далее проводят анализ, как указано в 10.3.4.2.
Из объединенных хлороформных экстрактов дважды реэкстрагируют никель 0,5 моль/дм3 раствором соляной кислоты, применяя каждый раз по 10 см3 кислоты и встряхивая содержимое воронки в течение 1 мин. Солянокислые растворы сливают в стакан вместимостью 50 см3, нагревают до кипения, охлаждают и переливают в мерную колбу вместимостью 100 см3. Прибавляют последовательно 2 см3 бромной воды или 2 см3 спиртового раствора йода, 2 см3 раствора диметилглиоксима и 20 см3 раствора углекислого натрия, доливают раствор водой до метки и перемешивают. Через 10 мин измеряют оптическую плотность раствора при длине волны 450 нм в кювете толщиной поглощающего свет слоя 50 мм.
Раствором сравнения при измерении оптической плотности раствора служит вода.
Одновременно проводят два контрольных опыта так, как указано в 10.3.4.2. Экстракцию, реэкстракцию и измерение оптической плотности раствора выполняют так, как указано выше.
Среднее значение оптической плотности раствора контрольного опыта вычитают из значения оптической плотности анализируемого раствора.
Массу никеля в растворе устанавливают по градуировочному графику, построенному, как указано в 10.3.3.6.
10.3.4.5 Определение никеля при массовой доле его от 0,05% до 0,5%
Навеску меди массой 1,0000 г растворяют и упаривают, как указано в 10.3.4.2.
Охлажденный раствор переводят в мерную колбу вместимостью 100 см3, доливают водой до метки и перемешивают. 10 см3 раствора переводят пипеткой в стакан вместимостью 100 см3, прибавляют раствор аммиака, разбавленный 1:1, до появления осадка гидроксидов, а затем по каплям соляную кислоту, разбавленную 1:3, до растворения осадка. К раствору приливают 3 см3 тартратного буферного раствора, 3 см3 ацетатного буферного раствора, 2,5 см3 серноватистокислого натрия, перемешивают и устанавливают величину рН (6,5 ±0,3). Раствор переносят в делительную воронку вместимостью 100 см3 и далее проводят анализ, как указано в 10.3.4.4.
Одновременно проводят два контрольных опыта. Для этого в стакан вместимостью 100 см3 помещают 1 см3 азотной кислоты, разбавленной 1:1, и выпаривают досуха. К остатку прибавляют 1 см3 0,5 моль/дм3 раствора соляной кислоты, 3 см3 тартратного буферного раствора, 3 см3 ацетатного буферного раствора, 2,5 см3 серноватистокислого натрия, перемешивают и устанавливают рН (6,5 ±0,3). Раствор переносят в делительную воронку вместимостью 100 см3 и далее анализ проводят, как указано в 10.3.4.4.
Среднее значение оптической плотности раствора контрольного опыта вычитают из значения оптической плотности анализируемого раствора.
Массу никеля в растворе устанавливают по градуировочному графику, построенному, как указано в 10.3.3.7.
10.3.4.6 Допускается использование электролита после выделения меди в соответствии с разделом 5.
В электролит добавляют 5 см3 раствора серной кислоты, разбавленной 1:1, и выпаривают до выделения паров серной кислоты, охлаждают, приливают от 5 до 10 см3 воды и выпаривание повторяют.
К охлажденному остатку приливают от 30 до 50 см3 воды, кипятят в течение 5 - 7 мин, охлаждают и, если есть нерастворимый остаток, фильтруют на фильтр «синяя лента», в конус которого вложено немного фильтробумажной массы, собирая фильтрат в мерную колбу вместимостью 100, 200 или 500 см3 в зависимости от массовой доли никеля. Остаток на фильтре промывают 4 - 5 раз водой и фильтр отбрасывают. Фильтрат в мерной колбе разбавляют водой до метки и перемешивают.
Аликвотную часть, содержащую никель в интервале от 0,005 до 0,05 мг, помещают в мерную колбу вместимостью 50 см3, приливают 2,5 см3 раствора сегнетовой соли, 7,5 см3 раствора гидроксида натрия с массовой концентрацией 100 г/дм3, 10 см3 раствора надсернокислого аммония и 10 см3 раствора диметилглиоксима в растворе гидроксида натрия с массовой концентрацией 80 г/дм3, затем разбавляют водой до метки и перемешивают. По истечении от 10 до 20 мин измеряют оптическую плотность раствора, как указано в 10.3.4.2.
Массу никеля определяют по градуировочному графику, построенному, как указано в 10.3.3.8.
10.4 Атомно-абсорбционный метод
10.4.1 Средства измерений, вспомогательные устройства, материалы, растворы
При выполнении анализа применяют следующие средства измерений, вспомогательные устройства:
- спектрофотометр атомно-абсорбционный с источником излучения на никель;
- компрессор воздушный;
- весы лабораторные специального класса точности по ГОСТ 24104;
- пипетки не ниже 2-го класса точности по ГОСТ 29169 и ГОСТ 29227;
- колбы мерные 2-100-2, 2-1000-2 по ГОСТ 1770;
- стаканы В-1-250 ТХС по ГОСТ 25336;
- колбы Кн-1-250- 19/26 по ТХС ГОСТ 25336.
При выполнении анализа применяют следующие материалы, растворы:
- ацетилен по ГОСТ 5457;
- воду дистиллированную по ГОСТ 6709;
- кислоту азотную по ГОСТ 4461, разбавленную 1:1 и 3:2;
- кислоту соляную по ГОСТ 3118;
- пропан-бутан по ГОСТ 20448;
- медь, СО для спектрального анализа, содержащий 2•10-4 % никеля, или электролитная медь с установленным содержанием никеля, раствор меди массовой концентрации 100 г/дм3;
- никель по ГОСТ 849;
- растворы никеля известной концентрации.
10.4.2 Метод анализа
Метод основан на измерении при длине волны 232,0, 323,3 и 352,4 нм поглощения резонансных линий никеля при введении анализируемого раствора в пламя ацетилен-воздух или пропан-бутан-воздух. При массовой доле никеля до 0,004% предварительно проводят концентрирование экстракцией хлороформом комплекса никеля с диметилглиоксимом.
10.4.3 Подготовка к выполнению анализа
10.4.3.1 При приготовлении раствора меди массовой концентрации 100 г/дм3 навеску 10 г стандартного образца меди помещают в коническую колбу вместимостью 250 см3 и растворяют при нагревании в 70 см3 азотной кислоты, разбавленной 1:1. Раствор упаривают для удаления основной массы кислоты, охлаждают и переносят в мерную колбу вместимостью 100 см3, доливают до метки водой и перемешивают.
10 см3 раствора меди содержит 2 мкг никеля.
10.4.3.2 Для построения градуировочных графиков готовят растворы никеля известной концентрации.
При приготовлении раствора А массовой концентрации никеля 0,1 мг/см3 навеску 0,100 г никеля растворяют в 10 см3 азотной кислоты, разбавленной 3:2, раствор переводят в мерную колбу вместимостью 1000 см3, доливают водой до метки и перемешивают.
При приготовлении раствора Б массовой концентрации никеля 0,01 мг/см310 см3 раствора А помещают в мерную колбу вместимостью 100 см3, доливают до метки водой и перемешивают.
10.4.3.3 Построение градуировочных графиков
Построение градуировочного графика при массовой доле никеля от 0,0005% до 0,004%
В семь стаканов вместимостью 250 см3 каждый помещают по 1,0000 г СО меди и далее продолжают растворение, как указано в 10.3.4.2. Затем к содержимому стаканов добавляют 0; 0,2; 0,5; 1,0; 2,0; 3,0 и 4,0 см3 раствора Б, что соответствует 2,0; 4,0; 7,0; 12,0; 22,0; 32,0; 42 мкг никеля, растворы выпаривают до объема 3 см3, остаток растворяют в 10 см3 воды и далее продолжают анализ, как в 10.3.4.2 или 10.3.4.3.
Измеряют поглощение линии никеля при длине волны 232,0 нм и по полученным данным строят градуировочный график.
Построение градуировочного графика при массовой доле никеля от 0,004% до 0,05%
В мерные колбы вместимостью 100 см3 каждая помещают 0; 1; 2; 5; 8; 10; 20 и 50 см3 раствора Б, по 10 см3 раствора меди, доливают водой до метки и перемешивают, измеряют поглощение при длине волны 232,0 нм.
Полученные растворы содержат 2,12, 22, 52, 82, 102, 202 и 502 мкг никеля.
Построение градуировочного графика при массовой доле никеля от 0,05% до 0,4%
В мерные колбы вместимостью 100 см3 каждая помещают 2; 5; 10; 20 и 40 см3 раствора А, что соответствует 0,2; 0,5; 1,0; 2,0 и 4,0 мг никеля, раствор доливают водой до метки и перемешивают, измеряют поглощение при длине волны 323,3 или 352,4 нм.
По полученным значениям оптических плотностей растворов и соответствующим им содержаниям никеля строят градуировочные графики.
10.4.4 Выполнение анализа
10.4.4.1 Общие требования к методам анализа и требования безопасности при выполнении анализов - в соответствии с разделами 3 и 4.
10.4.4.2 Навеску меди массой 1,0000 г помещают в коническую колбу вместимостью 100 см3 и растворяют при нагревании в 10 см3 азотной кислоты, разбавленной 1:1. Если после растворения меди остается нерастворимый осадок черного цвета, к раствору приливают от 1 до 2 см3 соляной кислоты и раствор упаривают до влажных солей. Содержимое колбы охлаждают, прибавляют 10 см3 воды и нагревают до растворения солей. Раствор охлаждают, переводят в мерную колбу вместимостью 100 см3, доливают до метки водой и перемешивают. Полученный раствор меди распыляют в пламя ацетилен-воздух или пропан-бутан-воздух атомно-абсорбционного спектрофотометра и измеряют абсорбцию при длине волны 232,0 или 323,3, или 352,4 нм.
Одновременно проводят контрольный опыт со всеми применяемыми реактивами. Значение оптической плотности раствора контрольного опыта вычитают из значения оптической плотности анализируемого раствора.
Массу никеля в растворе определяют по градуировочным графикам.
Допускается для определения массовой доли никеля использовать метод добавок.
10.4.4.3 При определении никеля массовой долей от 0,0005% до 0,004% растворение навески, выделение никеля, экстракцию и реэкстракцию проводят в соответствии с 10.3.4.2 и 10.3.4.3.
Реэкстракты помещают в мерную колбу вместимостью 25 см3, разбавляют водой до метки и перемешивают. Измеряют поглощение линии никеля при длине волны 232,0 нм одновременно с растворами контрольного опыта и растворами для построения градуировочного графика.
Допускается определение в анализируемом растворе цинка (от 0,0005% до 0,006%), свинца (от 0,005% до 0,06%), кобальта (от 0,005% до 0,06%).
10.4.4.4 Допускается использование электролита после выделения меди в соответствии с разделом 5. Для этого часть электролита (в зависимости от массовой доли никеля) помещают в стакан (или колбу) вместимостью 100 см3 и распыляют раствор в пламени ацетилен-воздух или пропан-бутан-воздух при длинах волн 232,0, 323,3 или 352,4 нм в зависимости от концентрации никеля в анализируемом растворе.
10.5 Обработка результатов анализа
10.5.1 Массовую долю никеля X, %, при использовании фотометрического метода вычисляют по формулам:
- при массовой доле никеля от 0,0005% до 0,05%
|
|
(17) |
- при массовой доле никеля от 0,05% до 0,4%
|
|
(18) |
где т1 - масса никеля, найденная по градуировочному графику, мкг;
V - объем анализируемого раствора, см3;
V1 - объем аликвотной части анализируемого раствора, см3;
т - масса навески меди, г.
10.5.2 Массовую долю никеля X, %, при использовании атомно-абсорбционного метода вычисляют по формуле
|
|
(19) |
где т1 - масса никеля, найденная по градуировочному графику, мкг;
т - масса навески меди, г.
За результат анализа принимают среднеарифметическое значение двух параллельных определений при условии, что абсолютная разность между ними в условиях повторяемости не превышает значений (при доверительной вероятности Р = 0,95) предела повторяемости г, приведенных в таблице 8.
Если расхождение между результатами параллельных определений превышает значение предела повторяемости, выполняют процедуры, изложенные в стандарте [2] (подпункт 5.2.2.1).
10.6 Контроль точности результатов анализа
Контроль точности результатов анализа - по 3.14.
10.7 Оформление результатов анализа
Результаты анализа оформляют в соответствии с 3.15, значения погрешности результатов анализа А приведены в таблице 8.
11 Метод определения массовой доли свинца
11.1 Область применения
Настоящий раздел устанавливает определение массовой доли свинца в меди в диапазоне от 0,0005% до 0,06% атомно-абсорбционным методом.
11.2 Требования к погрешности анализа
Погрешность результатов анализа массовой доли свинца, значения пределов повторяемости и воспроизводимости для доверительной вероятности Р = 0,95 должны соответствовать приведенным в таблице 9.
В процентах
|
Диапазон массовой доли свинца |
Погрешность результатов анализа ±∆ |
Предел |
|
|
повторяемости r (n=2) |
воспроизводимости R |
||
|
От 0,0005 до 0,0010 включ. |
0,0002 |
0,0002 |
0,0003 |
|
Св. 0,0010 » 0,0030 » |
0,0004 |
0,0004 |
0,0006 |
|
» 0,0030 » 0,0100 » |
0,0007 |
0,0008 |
0,0010 |
|
» 0,010 » 0,030 » |
0,003 |
0,002 |
0,004 |
|
» 0,030 » 0,060 » |
0,004 |
0,004 |
0,006 |
11.3 Средства измерений, вспомогательные устройства, материалы, растворы
При выполнении анализа применяют следующие средства измерений, вспомогательные устройства:
- спектрофотометр атомно-абсорбционный с источником излучения на свинец;
- компрессор воздушный;
- весы лабораторные специального класса точности по ГОСТ 24104;
- пипетки не ниже 2-го класса точности по ГОСТ 29169 и ГОСТ 29227;
- колбы мерные 2-25-2, 2-50-2, 2-100-2 по ГОСТ 1770;
- стаканы В-1-400 ТХС по ГОСТ 25336;
- колбы Кн-1-100-19/26 ТХС, Кн-1-250-19/26 ТХС по ГОСТ 25336.
При выполнении анализа применяют следующие материалы, растворы:
- ацетилен по ГОСТ 5457;
- кислоту азотную по ГОСТ 4461 и разбавленную 1:1;
- медь по ГОСТ 859 и раствор меди массовой концентрации 100 г/дм3;
- свинец высокой чистоты по ГОСТ 22861;
- растворы свинца известной концентрации;
- кислоту соляную по ГОСТ 3118 и разбавленную 1:1;
- аммиак водный по ГОСТ 3760 и раствор 1:99;
- железо по ГОСТ 9849, азотнокислый раствор массовой концентрации 25 г/дм3;
- универсальную индикаторную бумагу по [14];
- фильтры обеззоленные по [21].
11.4 Метод анализа
Метод анализа основан на растворении пробы в азотной кислоте и измерении при длине волны 283,3 нм поглощения резонансного излучения атомами свинца, образующимися в результата атомизации при введении солянокислого или азотнокислого растворов в пламя ацетилен-воздух.
11.5 Подготовка к выполнению анализа
11.5.1 При приготовлении раствора меди массовой концентрации 100 г/дм3 навеску 10,0 г меди помещают в коническую колбу вместимостью 250 см3, приливают 70 см3 азотной кислоты, разбавленной 1:1, и нагревают до растворения. Раствор переводят в мерную колбу вместимостью 100 см3, доливают до метки водой и перемешивают.
1 см3 раствора содержит 0,1 г меди.
11.5.2 Для построения градуировочных графиков готовят растворы свинца известной концентрации.
При приготовлении раствора А массовой концентрации свинца 1 мг/см3 навеску 1,0 г свинца помещают в коническую колбу вместимостью 100 см3 и растворяют в 10 см3 азотной кислоты, разбавленной 1:1. Раствор переводят в мерную колбу вместимостью 1000 см3, доливают водой до метки и перемешивают.
При приготовлении раствора Б массовой концентрации свинца 0,05 мг/см3 50 см3 раствора А помещают в мерную колбу вместимостью 1000 см3, доливают до метки водой и перемешивают.
11.5.3 При приготовлении раствора азотнокислого железа массовой концентрации 25 г/дм3 навеску 2,5 г железа растворяют в 50 см3 азотной кислоты, разбавленной 1:1. Раствор переводят в мерную колбу вместимостью 100 см3, доливают водой до метки и перемешивают.
11.5.4 Построение градуировочного графика при массовой доле свинца от 0,005% до 0,06%
В мерные колбы вместимостью 100 см3 каждая помещают 0; 2,0; 5,0; 10,0; 20,0 и 30,0 см3 раствора Б, что соответствует 0; 0,1; 0,25; 0,5; 1,0 и 1,5 мг свинца, прибавляют по 10 см3 раствора меди, доливают водой до метки и перемешивают.
Измеряют абсорбцию приготовленных растворов, как указано в 11.6.2. По полученным значениям оптических плотностей растворов и соответствующим им значениям содержания свинца строят градуировочный график.
11.5.5 Построение градуировочного графика при массовой доле свинца от 0,0005% до 0,005%
Для построения градуировочного графика в стаканы вместимостью 400 см3 каждый помещают по 5,0 г меди и растворяют в соответствии с 11.6.2; приливают от 200 до 250 см3 воды и 5 см3 азотнокислого раствора железа, добавляют 0; 0,5; 1,0; 2,0; 4,0 и 6,0 см3 раствора Б, что соответствует 0; 0,025; 0,05; 0,1; 0,2 и 0,3 мг свинца, и далее анализ продолжают в соответствии с 11.6.3. По полученным значениям оптической плотности и соответствующим им концентрациям растворов строят градуировочный график.
11.6 Выполнение анализа
11.6.1 Общие требования к методам анализа и требования безопасности при выполнении анализов--в соответствии с разделами 3 и 4.
11.6.2 При определении свинца массовой долей свыше 0,005% навеску меди массой 2,0000 г помещают в коническую колбу вместимостью 100 см3 и растворяют в 20 - 25 см3 азотной кислоты, разбавленной 1:1. Раствор переводят в мерную колбу вместимостью 100 см3, доливают до метки водой и перемешивают. Полученный раствор меди распыляют в пламя атомно-абсорбционного спектрофотометра и измеряют абсорбцию при длине волны 283,3 нм. Одновременно проводят контрольный опыт со всеми применяемыми реактивами.
Значение оптической плотности раствора контрольного опыта вычитают из значения оптической плотности анализируемого раствора.
Массу свинца в растворе определяют по градуировочному графику.
Допускается для определения массовой доли свинца использовать метод добавок.
11.6.3 При массовой доле свинца до 0,005% навеску меди массой 5,0000 г помещают в стакан вместимостью 400 см3 и растворяют в 25 см3 азотной кислоты. Нагревают до прекращения выделения оксидов азота. Затем приливают 250 см3 воды и 5 см3 раствора железа, нагревают до температуры 60°С. Приливают аммиак в таком количестве, чтобы вся медь перешла в аммиачный комплекс (синяя окраска раствора), и еще 5 см3. Нагревают раствор при температуре от 60°С до 70°С до коагуляции осадка. Фильтруют через фильтр средней плотности «белая лента» и промывают 3 - 4 раза горячим раствором аммиака 1:99. Растворяют осадок на фильтре в 10 см3 горячей соляной кислоты, разбавленной 1:1, собирая фильтрат в стакан, где проводили осаждение. Фильтр промывают горячей водой до нейтральной реакции фильтрата (проверка по универсальной индикаторной бумаге). Раствор после охлаждения помещают в мерную колбу вместимостью 25 или 50 см3, разбавляют водой до метки и перемешивают.
Измеряют поглощение линии свинца, распыляя раствор в пламени ацетилен-воздух, при длине волны 283,3 нм одновременно с раствором контрольного опыта и растворами для построения градуировочного графика.
Допускается определение в анализируемом растворе висмута (от 0,0003% до 0,005%), олова (от 0,01% до 0,06%) и сурьмы (от 0,0005% до 0,02%).
Массу свинца определяют по градуировочному графику.
11.7 Обработка результатов
11.7.1 Массовую долю свинца X, %, вычисляют по формуле
|
|
(20) |
где m1 - масса свинца, найденная по градуировочному графику, мкг;
т - масса навески меди, г.
11.7.2 За результат анализа принимают среднеарифметическое значение двух параллельных определений при условии, что абсолютная разность между ними в условиях повторяемости не превышает значений (при доверительной вероятности Р = 0,95) предела повторяемости г, приведенных в таблице 9.
Если расхождение между результатами параллельных определений превышает значение предела повторяемости, выполняют процедуры, изложенные в стандарте [2] (подпункт 5.2.2.1).
11.8 Контроль точности результатов анализа
Контроль точности результатов анализа - по 3.14.
11.9 Оформление результатов анализа
Результаты анализа оформляют в соответствии с 3.15, значения погрешности результатов анализа ∆ приведены в таблице 9.
12 Метод определения массовой доли олова
12.1 Область применения
Настоящий раздел устанавливает определение массовой доли олова в меди в диапазоне от 0,0005% до 0,08% фотометрическим методом.
Метод не распространяется на определение массовой доли олова в меди по ГОСТ 859 марок М00к и М00б.
12.2 Требования к погрешности анализа
Погрешность результатов анализа массовой доли олова, значения пределов повторяемости и воспроизводимости для доверительной вероятности Р = 0,95 должны соответствовать приведенным в таблице 10.
В процентах
|
Диапазон массовой доли олова |
Погрешность результатов анализа ±∆ |
Предел |
|
|
повторяемости r (n=2) |
воспроизводимости R |
||
|
От 0,0005 до 0,0025 включ. |
0,0004 |
0,0003 |
0,0005 |
|
Св. 0,0025 » 0,0060 » |
0,0007 |
0,0007 |
0,0010 |
|
» 0,006 » 0,020 » |
0,002 |
0,002 |
0,003 |
|
» 0,020 » 0,080 » |
0,004 |
0,004 |
0,006 |
12.3 Средства измерений, вспомогательные устройства, материалы, растворы
При выполнении анализа применяют следующие средства измерений, вспомогательные устройства:
- фотометр фотоэлектрический или спектрофотометр со всеми принадлежностями, обеспечивающими проведение измерений при длине волны 510 нм;
- весы лабораторные специального класса точности по ГОСТ 24104;
- пипетки не ниже 2-го класса точности по ГОСТ 29169 и ГОСТ 29227;
- колбы мерные 2-25-2, 2-100-2, 2-500-2, 2-1000-2 по ГОСТ 1770;
- стаканы В-1-100 ТХС, В-1-150 ТХС, В-1-400 ТХС по ГОСТ 25336;
- воронки ВД-1-100 ХС, ВД-1-250 ХС по ГОСТ 25336.
При выполнении анализа применяют следующие материалы, растворы:
- кислоту серную по ГОСТ 4204, и разбавленную 1:1 и 1:9;
- кислоту соляную по ГОСТ 3118;
- кислоту азотную по ГОСТ 4461;
- кислоту хлорную по [23];
- водорода пероксид по ГОСТ 10929 и раствор 1:9;
- тиомочевину по ГОСТ 6344, 10%-ный раствор;
- N-нитрозо-N-фенилгидроксиламин аммонийную соль, раствор массовой концентрации 20 г/дм3, свежеприготовленный;
- хлороформ по ГОСТ 20015, перегнанный;
- кислоту винную по ГОСТ 5817, 50%-ный раствор;
- кислоту аскорбиновую, 10%-ный раствор, свежеприготовленный;
- аммиак водный по ГОСТ 3760 и разбавленный 1:1, 1:50;
- бром по ГОСТ 4109, насыщенный водный раствор (бромная вода);
- квасцы железоаммонийные по [24], раствор массовой концентрации 17,5 г/дм3;
- желатин по ГОСТ 11293, раствор массовой концентрации 5 г/дм3;
- спирт этиловый по ГОСТ 18300;
- фенилфлуорон (2,3,7-тригидрокси-9-фенил-6-флуорон), 0,03%-ный спиртовый раствор, содержащий 1 см3 раствора серной кислоты в 100 см3;
- натрий хлористый по ГОСТ 4233;
- олово по ГОСТ 860, марка 01;
- растворы олова известной концентрации;
- бумагу индикаторную «конго».
12.4 Метод анализа
Метод основан на образовании комплексного соединения олова (IV) с фенилфлуороном после отделения олова от мешающих элементов экстракцией хлороформом в виде купфероната или соосаждением с гидроксидом железа (III). Оптическую плотность раствора измеряют при длине волны 510 нм.
12.5 Подготовка к выполнению анализа
12.5.1 При приготовлении раствора желатина массовой концентрации 5 г/дм3 навеску 0,5 г желатина помещают в стакан вместимостью 150 см3, прибавляют 20 см3 воды и оставляют для набухания. Затем прибавляют 70 см3 воды, нагретой до температуры от 70°С до 80°С, нагревают до кипения и охлаждают. Раствор доливают до 100 см3 водой и перемешивают. Раствор готовят непосредственно перед употреблением.
12.5.2 Для построения градуировочного графика готовят растворы олова известной концентрации.
При приготовлении раствора А массовой концентрации олова 0,1 мг/см3 навеску 0,1 г олова растворяют в 10 см3 серной кислоты при нагревании. Раствор помещают в мерную колбу вместимостью 1000 см3, доливают до метки серной кислотой, разбавленной 1:9, и перемешивают.
При приготовлении раствора Б массовой концентрации олова 0,01 мг/см310 см3 раствора А помещают в мерную колбу вместимостью 100 см3, доливают до метки серной кислотой, разбавленной 1:9, и перемешивают. Раствор готовят непосредственно перед употреблением.
12.5.3 Построение градуировочного графика
В мерные колбы вместимостью 25 см3 каждая помещают 0; 0,25; 0,5; 1,0; 2,0; 3,0 и 4,0 см3 раствора Б, что соответствует 0; 2,5; 5; 10; 20; 30 и 40 мкг олова. Во все колбы прибавляют 1,0 см3 раствора железоаммонийных квасцов, 0,3 см3 раствора винной кислоты, нейтрализуют раствором аммиака 1:1 по индикаторной бумаге «конго» и далее поступают, как указано в 12.6.3.
Раствором сравнения служит раствор, не содержащий раствор Б.
По полученным значениям оптических плотностей и соответствующим им значениям содержания олова строят градуировочный график.
12.6 Выполнение анализа
12.6.1 Общие требования к методам анализа и требования безопасности при выполнении анализов- в соответствии с разделами 3 и 4.
12.6.2 Навеску меди массой 5,0000 г растворяют в 75 см3 соляной кислоты в присутствии 1 г хлористого натрия. Постепенно прибавляют 20 см3 пероксида водорода, не допуская бурной реакции. Избыток пероксида водорода удаляют кипячением. Раствор охлаждают, помещают в мерную колбу вместимостью 500 см3, доливают водой до метки и перемешивают. Содержание олова в полученном растворе определяют, как указано в 12.6.3 и 12.6.4.
12 6.3 50 см3 раствора при массовой доле олова от 0,0005% до 0,005% или 5 см3 раствора при массовой доле олова от 0,005% до 0,08% помещают в делительную воронку вместимостью 250 см3. В случае применения 5 см3 раствора доливают раствором соляной кислоты до объема 50 см3. К раствору в делительной воронке прибавляют 50 см3 тиомочевины и перемешивают. Затем прибавляют 5 см3 раствора N-митрозо-N-фенилгидроксиламин аммонийной соли, перемешивают и через 2 мин экстрагируют олово-купфероновый комплекс 10 см3 хлороформа, встряхивая содержимое в течение 2 мин. Органический слой помещают в делительную воронку вместимостью 100 см3. Экстракцию проводят два раза. Объединенные экстракты промывают дважды 5 см3 серной кислотой, разбавленной 1:9, Экстракт помещают в стакан вместимостью 100 см3, добавляют 5 см3 серной кислоты, разбавленной 1:1,5 см3 раствора азотной кислоты, 2 см3 хлорной кислоты и раствор упаривают на песчаной бане до объема 1 см3.
После охлаждения добавляют 2 см3 воды, 1,0 см3 раствора железоаммонийных квасцов и 0,3 см3 раствора винной кислоты. Раствор нейтрализуют раствором аммиака 1:1 по индикаторной бумаге «конго» и помещают в мерную колбу вместимостью 25 см3. К раствору добавляют 1,6 см3 серной кислоты, разбавленной 1:1, и бромную воду до желтой окраски.
Через 5 мин добавляют по каплям раствор аскорбиновой кислоты до обесцвечивания раствора и избыток 1 см3. Через 5 мин добавляют 2,5 см3 раствора желатина, 5,0 см3 раствора фенилфлуорона и доливают раствор водой до метки. После прибавления каждого реагента раствор перемешивают. Через 30 мин измеряют оптическую плотность раствора при длине волны 510 нм. Раствором сравнения служит раствор контрольного опыта.
Массу олова устанавливают по градуировочному графику, построенному, как указано в 12.5.3.
12.6.4 Для отделения олова соосаждением с гидроксидом железа помещают 50 см3 раствора, полученного в 12.6.2, при массовой доле олова до 0,005% или 5 см3 раствора при массовой доле олова выше 0,005% в стакан вместимостью 400 см3 и доливают водой до 200 см3. К раствору прибавляют 1 см3 раствора железоаммонийных квасцов и нагревают до температуры от 60°С до 70°С. При постоянном перемешивании прибавляют аммиак в таком количестве, чтобы вся медь перешла в комплексное соединение (синяя окраска раствора), и в избытке 5 см3. Через 15 мин осадок фильтруют на фильтр средней плотности «белая лента». Стенки стакана дважды обмывают горячим раствором аммиака 1:50.
Фильтр с осадком промывают 5 - 6 раз этим же раствором аммиака. Осадок с фильтра смывают в стакан, в котором проводили осаждение гидроксидов, прибавляют 10 см3 серной кислоты, разбавленной 1:1, 150 см3 воды и переосаждают гидроксиды. Осадок фильтруют на тот же фильтр и промывают 2 раза горячим раствором аммиака 1:50. Осадок гидроксидов смывают с фильтра в стакан, в котором проводили переосаждение, фильтр промывают несколько раз 20 см3 горячей серной кислоты, разбавленной 1:9, собирая раствор в стакан с осадком. К раствору прибавляют 5 см3 серной кислоты, разбавленной 1:1,5 см3 азотной кислоты, 2 см3 хлорной кислоты. Растворы упаривают на песчаной бане до объема 1 см3.
После охлаждения добавляют 2 см3 воды и 0,3 см3 раствора винной кислоты, нейтрализуют раствором аммиака 1:1 по индикаторной бумаге «конго» и далее поступают, как указано в 12.6.3.
12.7 Обработка результатов анализа
12.7.1 Массовую долю олова X, %, вычисляют по формуле
|
|
(21) |
где m1 - масса олова, найденная по градуировочному графику, мкг;
V - объем анализируемого раствора, см3;
т - масса навески меди, г;
V1 - объем аликвотной части раствора, см3.
12.7.2 За результат анализа принимают среднеарифметическое значение двух параллельных определений при условии, что абсолютная разность между ними в условиях повторяемости не превышает значений (при доверительной вероятности Р = 0,95) предела повторяемости г, приведенных в таблице 10.
Если расхождение между результатами параллельных определений превышает значение предела повторяемости, выполняют процедуры, изложенные в стандарте [2] (подпункт 5.2.2.1).
12.8 Контроль точности результатов анализа
Контроль точности результатов анализа - в соответствии с 3.14.
12.9 Оформление результатов анализа
Результаты анализа оформляют в соответствии с 3.15, значения погрешности результатов анализа ∆ приведены в таблице 10.
13 Методы определения массовой доли серебра
13.1 Область применения
Настоящий раздел устанавливает определение массовой доли серебра в меди в диапазоне от 0,0010% до 0,02% фотометрическим и атомно-абсорбционным методами.
13.2 Требования к погрешности анализа
Погрешность результатов анализа массовой доли серебра, значения пределов повторяемости и воспроизводимости для доверительной вероятности Р = 0,95 должны соответствовать приведенным в таблице 11.
В процентах
|
Диапазон массовой доли серебра |
Погрешность результатов анализа ±∆ |
Предел |
|
|
повторяемости r (n=3) |
воспроизводимости R |
||
|
От 0,0010 до 0,0020 включ. |
0,0005 |
0,0006 |
0,0007 |
|
Св. 0,0020 » 0,0040 » |
0,0007 |
0,0008 |
0,0010 |
|
» 0,004 » 0,008 » |
0,001 |
0,001 |
0,002 |
|
» 0,008 » 0,020 » |
0,002 |
0,002 |
0,003 |
|
» 0,020 |
0,004 |
0,003 |
0,005 |
13.3 Фотометрический метод
13.3.1 Средства измерений, вспомогательные устройства, материалы, растворы
При выполнении анализа применяют следующие средства измерений, вспомогательные устройства:
- фотометр фотоэлектрический или спектрофотометр со всеми принадлежностями, обеспечивающие проведение измерений при длине волны 440 нм;
- весы лабораторные специального класса точности по ГОСТ 24104;
- пипетки не ниже 2-го класса точности по ГОСТ 29169 и ГОСТ 29227;
- колбы мерные 2-100-2, 2-500-2, 2-1000-2 по ГОСТ 1770;
- стаканы В-1-50 ТС по ГОСТ 25336;
- воронку ВД-1-250 ХС по ГОСТ 25336.
При выполнении анализа применяют следующие материалы, растворы:
- кислоту азотную по ГОСТ 4461, разбавленную 1:1;
- кислоту серную по ГОСТ 4204, разбавленную 1:1;
- кислоту соляную по ГОСТ 3118;
- аммиак водный по ГОСТ 3760, разбавленный 1:5;
- аммоний уксуснокислый по ГОСТ 3117, раствор массовой концентрации 200 г/дм3;
- соль динатриевую этилендиамин-N, N, N', N'-тетрауксусной кислоты 2-водную (трилон Б) по ГОСТ 10652, 0,1 М раствор;
- кислоту уксусную по ГОСТ 61;
- медь (II) сернокислую 5-водную по ГОСТ 4165;
- натрия N, N-диэтилдитиокарбамат 3-водный по ГОСТ 8864, раствор массовой концентрации 100 г/дм3;
- натрий уксуснокислый 3-водный по ГОСТ 199;
- толуол по ГОСТ 5789;
- бумагу индикаторную, универсальную по [14];
- фильтры обеззоленные по [21] или другие средней плотности;
- диэтилдитиокарбамат меди, раствор;
- ацетатный буферный раствор с рН 4;
- серебро азотнокислое по ГОСТ 1277;
- аммоний азотнокислый по ГОСТ 22867;
- растворы серебра известной концентрации.
13.3.2 Метод анализа
Метод основан на реакции обесцвечивания диэтилдитиокарбамата меди под воздействием ионов серебра в ацетатном буферном растворе при рН 4,0. Оптическую плотность раствора измеряют при длине волны 440 нм.
Мешающее влияние меди устраняют при помощи трилона Б.
13.3.3 Подготовка к выполнению анализа
13.3.3.1 При приготовлении раствора трилона Б молярной концентрации 0,1 М навеску 37,22 г соли динатриевой этилендиамин-N, N, N', N'-тетрауксусной кислоты 2-водной растворяют в 1 дм3 воды.
13.3.3.2 При приготовлении раствора диэтилдитиокарбамата меди навеску сернокислой меди массой 0,04 г помещают в стакан вместимостью 50 см3, растворяют в 40 см3 воды и приливают аммиак до рН 9 (контролируя по индикаторной бумаге). Полученный раствор переносят в делительную воронку вместимостью 250 см3, добавляют 4,5 см3 диэтилдитиокарбамата натрия, 20 см3 толуола и экстрагируют в течение 2 мин. После разделения слоев водный слой помещают в другую делительную воронку и повторяют экстракцию с 10 см3 толуола до тех пор, пока толуольный слой не станет бесцветным. Все толуольные вытяжки помещают в мерную колбу вместимостью 500 см3, доливают водой до метки и перемешивают.
Хранят в склянке из темного стекла в темном месте. Это - основной раствор.
Для работы готовят разбавленный раствор. Непосредственно перед применением 15 см3 основного pact вора смешивают с 185 см3 толуола.
13.3.3.3 При приготовлении ацетатного буферного раствора с рН 4 в мерную колбу вместимостью 1000 см3 помещают 11,5 см3 уксусной кислоты, навеску 5,44 г уксуснокислого натрия, доливают водой до метки и перемешивают.
13.3.3.4 Для построения градуировочных графиков готовят растворы серебра известной концентрации.
При приготовлении раствора А массовой концентрации серебра 1 мг/см3 навеску 0,1574 г азотнокислого серебра помещают в мерную колбу вместимостью 100 см3, доливают водой до метки и перемешивают.
При приготовлении раствора Б массовой концентрации серебра 50 мкг/см3 5 см3 раствора А помещают в мерную колбу вместимостью 100 см3, доливают до метки водой и перемешивают.
При приготовлении раствора В массовой концентрации серебра 5 мкг/см3 10 см3 раствора Б помещают в мерную колбу вместимостью 100 см3, доливают до метки водой и перемешивают.
Все растворы серебра необходимо хранить в темном месте.
13.3.3.5 Построение градуировочного графика
В стаканы вместимостью 50 см3 каждый помещают 0; 1,0; 2,0; 4,0; 6,0 и 8,0 см3 раствора В, что соответствует 0; 5,0; 10,0; 20,0; 30,0 и 40,0 мкг серебра, прибавляют 2 см3 серной кислоты, разбавленной 1:1, и проводят все операции, как указано в 13.3.4.2.
По полученным значениям оптических плотностей растворов и соответствующим им значениям содержания серебра строят градуировочный график.
13.3.4 Выполнение анализа
13.3.4.1 Общие требования к методам анализа и требования безопасности при выполнении анализов - в соответствии с разделами 3 и 4.
13.3.4.2 Навеску массой 1,0000 г помещают в коническую колбу вместимостью 250 см3, прибавляют 20 см3 азотной кислоты, разбавленной 1:1. После окончания бурной реакции раствор нагревают до полного растворения меди. Раствор охлаждают, прибавляют 10 см3 серной кислоты, разбавленной 1:1, и выпаривают до выделения паров серной кислоты. Если раствор окрашен в желтый цвет (органика), к содержимому колбы добавляют азотнокислый аммоний (на кончике шпателя) и нагревают до обесцвечивания раствора. Содержимое колбы охлаждают, стенки колбы обмывают от 5 до 7 см3 воды, и нагревание продолжают до выделения паров серной кислоты. Эту операцию повторяют два раза. Объем остатка серной кислоты должен быть около 2 см3.
Содержимое колбы охлаждают, прибавляют 10 см3 воды и нагревают до кипения. Раствор охлаждают, нерастворимый осадок фильтруют на фильтр средней плотности, помещают фильтрат в стакан вместимостью 50 см3, фильтр промывают водой, небольшими порциями. Объем фильтрата и промывных вод не должен превышать 20 см3.
При массовой доле серебра менее 0,005% используют весь раствор; если массовая доля серебра более 0,005%, то фильтрат помещают в мерную колбу вместимостью 100 см3 и доливают до метки водой. Для дальнейшего анализа отбирают аликвотную часть.
Если при фильтровании выпадает осадок, то после выпаривания с серной кислотой остаток осторожно обмывают от 2 до 3 см3 воды, добавляют 2 см3 трилона Б, 10 см3 буферной ацетатной смеси и нейтрализуют раствор по универсальной индикаторной бумаге до рН 4. Фильтрат или аликвотную часть (от 5 до 20 см3) помещают в стакан вместимостью 50 см3 и выпаривают до 5 см3. После охлаждения приливают 2 см3 трилона Б, нейтрализуют раствор аммиаком до оранжево-желтой окраски (рН 4).
Переносят раствор в делительную воронку, приливают ацетатный буферный раствор до объема 20 см3 (метка на воронке), добавляют 10 см3 разбавленного раствора диэтилдитиокарбамата натрия и экстрагируют 2 мин. После расслоения органический слой сливают в сухую пробирку с притертой пробкой. По истечении от 20 до 30 мин измеряют оптическую плотность при длине волны 440 нм в кювете толщиной поглощающего свет слоя от 20 до 10 мм.
Раствором сравнения при измерении оптической плотности служит раствор, проведенный через все стадии анализа, начиная с осаждения, и содержащий все реагенты за исключением серебра. Массу серебра устанавливают по градуировочному графику, построенному, как указано в 13.3.3.5.
13.4 Атомно-абсорбционный метод
13.4.1 Средства измерений, вспомогательные устройства, материалы, растворы
При выполнении анализа применяют следующие средства измерений, вспомогательные устройства:
- спектрофотометр атомно-абсорбционный с источником излучения на серебро;
- весы лабораторные специального класса точности по ГОСТ 24104;
- пипетки не ниже 2-го класса точности по ГОСТ 29169 и ГОСТ 29227;
- колбы 2-100-2, 2-1000-2 по ГОСТ 1770;
- стаканы В-1-100 ТС по ГОСТ 25336;
- компрессор воздушный.
При выполнении анализа применяют следующие материалы, растворы:
- ацетилен по ГОСТ 5457;
- пропан-бутан по ГОСТ 20448;
- воду бидистиллированную;
- кислоту соляную по ГОСТ 3118;
- кислоту азотную по ГОСТ 4461, разбавленную 1:1; 2:1 и раствор молярной концентрации 0,1 моль/дм3;
- кислоту азотную особой чистоты по ГОСТ 11125;
- ртуть (II) азотнокислую 1-водную (нитрат ртути) по ГОСТ 4520, раствор массовой концентрации 25 г/дм3 в растворе азотной кислоты молярной концентрации 0,1 моль/дм3;
- медь, стандартные образцы для спектрального анализа, содержащие 25,5·10-4 % и 18,6·10-4 % серебра соответственно. Можно применять электролитную медь с установленным содержанием серебра; растворы меди массовой концентрации 100 г/дм3;
- серебро по ГОСТ 6836;
- растворы серебра известной концентрации.
13.4.2 Метод анализа
Метод основан на растворении пробы в азотной кислоте с добавлением соляной кислоты или нитрата ртути и измерении при длине волны 328,1 нм поглощения резонансного излучения атомами серебра, образующимися в результате атомизации при введении солянокислого или азотнокислого растворов серебра в пламя ацетилен-воздух.
13.4.3 Подготовка к выполнению анализа
13.4.3.1 Приготовление растворов меди массовой концентрации 100 г/дм3
При приготовлении раствора меди массовой концентрации 100 г/дм3 навеску 10,0 г СО растворяют при нагревании в 70 см3 азотной кислоты, разбавленной 1:1, и раствор упаривают для удаления основной массы кислоты. Затем прибавляют 10 см3 воды и нагревают до растворения солей. Раствор охлаждают и прибавляют 16 см3 соляной кислоты. Раствор переводят в мерную колбу вместимостью 100 см3, доливают до метки водой и перемешивают.
10 см3 раствора меди содержат: 25,5 мкг или 18,6 мкг серебра при использовании СО меди.
13.4.3.2 Для построения градуировочных графиков готовят растворы серебра известной концентрации.
При приготовлении раствора А массовой концентрации серебра 0,1 мг/см3 навеску 0,1 г серебра растворяют в 5 см3 азотной кислоты особой чистоты, раствор переводят в мерную колбу вместимостью 1000 см3, доливают до метки бидистиллированной водой и перемешивают. Раствор хранят в темном месте.
Такой же раствор может быть приготовлен следующим образом: навеску 0,1 г серебра растворяют в 25 см3 азотной кислоты, разбавленной 2:1, добавляют 5 см3 раствора нитрата ртути и нагревают до удаления оксидов азота. Раствор охлаждают, помещают в мерную колбу вместимостью 1000 см3, разбавляют до метки бидистиллированной водой и перемешивают. Раствор хранят в темном месте.
При приготовлении раствора Б массовой концентрации серебра 0,01 мг/см3 10 см3 раствора А помещают в мерную колбу вместимостью 100 см3 и перемешивают. Раствор хранят не более 5 ч.
Такой же раствор может быть приготовлен следующим образом: 10 см3 раствора А помещают в мерную колбу вместимостью 100 см3, приливают 5 см3 раствора нитрата ртути и 10 см3 азотной кислоты, разбавленной 1:1, разбавляют до метки бидистиллированной водой и перемешивают.
13.4.3.3 Построение градуировочного графика
В мерные колбы вместимостью 100 см3 каждая помещают 0; 1,0; 2,0; 3,0 и 4,0 см3 раствора Б, по 10 см3 азотнокислой меди, по 16 см3 соляной кислоты, доливают водой до метки и перемешивают. Полученные растворы содержат 0; 0,01; 0,02; 0,03; 0,04 мкг серебра.
Допускается приготовление растворов следующим образом. В мерные колбы вместимостью 100 см3 помещают по 10 см3 азотной кислоты, разбавленной 1:1, и по 2 см3 раствора нитрата ртути. Затем в колбы приливают последовательно 0; 1,0; 2,0; 3,0; и 4,0 см3 раствора Б, разбавляют водой до метки и перемешивают. Полученные растворы содержат 0; 0,01; 0,02; 0,03; 0,04 мкг серебра.
Допускается для построения градуировочного графика использовать СО меди с аттестованной массовой долей серебра.
При использовании раствора меди, приготовленного из СО, содержащего 18,6·10-4 % серебра растворы для градуировочного графика содержат 19, 29, 39, 49 и 59 мкг серебра или 26, 36, 46, 56 и 66 мкг серебра при использовании раствора меди, приготовленного из СО, содержащего 25,5·10-4 % серебра.
Далее проводят анализ, как указано в 13.3.4.2.
Приготовленные растворы анализируют на атомно-абсорбционном спектрофотометре. По полученным значениям оптических плотностей растворов и соответствующим им значениям содержания серебра строят градуировочный график.
13.4.4 Выполнение анализа
13.4.4.1 Общие требования к методам анализа и требования безопасности при выполнении анализов - в соответствии с разделами 3 и 4.
13.4.4.2 Навеску меди массой 1,0000 г растворяют при нагревании в 10 см3 азотной кислоты, разбавленной 1:1, и раствор упаривают до влажных солей. Затем прибавляют 10 см3 воды и нагревают до растворения солей. Раствор охлаждают и прибавляют 16 см3 соляной кислоты. Раствор переводят в мерную колбу вместимостью 100 см3, доливают до метки водой и перемешивают. Полученный раствор меди распыляют в пламя атомно-абсорбционного спектрофотометра и измеряют абсорбцию в пламени при длине волны 328,1 нм.
Одновременно проводят контрольный опыт со всеми применяемыми реактивами. Значение оптической плотности раствора контрольного опыта вычитают из значения оптической плотности анализируемого раствора.
Массу серебра в растворе устанавливают по градуировочному графику.
Допускается для определения массовой доли серебра использовать метод добавок.
13.4.4.3 Навеску меди массой 1,0 г помещают в стакан вместимостью 100 см3, приливают 10 см3 азотной кислоты, разбавленной 1:1, и 2 см3 раствора нитрата ртути. Нагревают до растворения навески. Раствор затем охлаждают, помещают в мерную колбу вместимостью 100 см3, разбавляют водой до метки и перемешивают.
Измеряют поглощение линии серебра в пламени ацетилен-воздух при длине волны 328,1 нм одновременно с растворами контрольного опыта.
Допускается одновременное определение в анализируемом растворе цинка (от 0,0005% до 0,006%), никеля (от 0,1% до 0,5%), свинца (от 0,005% до 0,06%) и железа (от 0,01% до 0,06%).
13.5 Обработка результатов анализа
13.5.1 Массовую долю серебра X, %, при использовании фотометрического и атомно-абсорбционного методов вычисляют по формуле
|
|
(22) |
где m1 - масса серебра, найденная по градуировочному графику, мкг;
т - масса навески меди, г.
13.5.2 За результат анализа принимают среднеарифметическое значение трех параллельных определений при условии, что абсолютная разность между максимальным и минимальным значениями в условиях повторяемости не превышает значений (при доверительной вероятности Р = 0,95) предела повторяемости г, приведенных в таблице 11.
Если расхождение между наибольшим и наименьшим результатами параллельных определений превышает значение предела повторяемости, выполняют процедуры, изложенные в стандарте [2] (подпункт 5.2.2.1).
13.6 Контроль точности результатов анализа
Контроль точности результатов анализа - в соответствии с 3.14.
13.7 Оформление результатов анализа
Результаты анализа оформляют в соответствии с 3.15, значения погрешности результатов анализа А приведены в таблице 11.
14 Методы определения массовой доли сурьмы
14.1 Область применения
В настоящем разделе установлено определение массовой доли сурьмы в меди в диапазоне от 0,0005% до 0,1% фотометрическим и атомно-абсорбционным методами.
14.2 Требования к погрешности анализа
Погрешность результатов анализа массовой доли сурьмы, значения пределов повторяемости и воспроизводимости для доверительной вероятности Р = 0,95 должны соответствовать приведенным в таблице 12.
В процентах
|
Диапазон массовой доли сурьмы |
Погрешность результатов анализа ±∆ |
Предел |
|
|
повторяемости r (n=2) |
воспроизводимости R |
||
|
От 0,0005 до 0,0010 включ. |
0,0002 |
0,0002 |
0,0003 |
|
Св. 0,0010 » 0,0030 » |
0,0004 |
0,0004 |
0,0006 |
|
» 0,003 » 0,010 » |
0,001 |
0,001 |
0,002 |
|
» 0,010 » 0,030 » |
0,003 |
0,002 |
0,004 |
|
» 0,030 » 0,100 » |
0,005 |
0,004 |
0,007 |
14.3 Фотометрический метод
14.3.1 Средства измерений, вспомогательные устройства, материалы, растворы
При выполнении анализа применяют следующие средства измерений, вспомогательные устройства:
-фотометр фотоэлектрический или спектрофотометр со всеми принадлежностями, обеспечивающими проведение измерений при длине волны 550 нм;
- центрифугу со всеми принадлежностями;
- весы лабораторные специального класса точности по ГОСТ 24104;
- пипетки не ниже 2-го класса точности по ГОСТ 29169 и ГОСТ 29227;
- колбы 2-100-2, 2-200-2, 2-500-2, 2-1000-2 по ГОСТ 1770;
- стаканы В-1-100 ТС, В-1-150 ТС, В-1 400 ТС по ГОСТ 25336;
- воронку ВД-1-250 ХС по ГОСТ 25336;
- дефлегматор 250-19/26-29/32 ТС по ГОСТ 25336;
- колбы Кн-1-250-19/26 ТС, Кн-1-1000-34/35 ТС по ГОСТ 25336;
- цилиндры 2-100-1 по ГОСТ 1770;
- бюретку 1-1-2-50-0,1 по ГОСТ 29251;
- стекло часовое.
При выполнении анализа применяют следующие материалы, растворы:
- кислоту соляную по ГОСТ 3118, раствор молярной концентрации 1 моль/дм3 и разбавленную 7:3;
- кислоту серную по ГОСТ 4204;
- гидроксиламин солянокислый по ГОСТ 5456, раствор массовой концентрации 10 г/дм3 в соляной кислоте молярной концентрации 1 моль/дм3 (устойчив в течение 3 суток);
- медь по ГОСТ 859, раствор;
- натрий сернокислый безводный по ГОСТ 4166;
- сульфит натрия безводный по ГОСТ 5644, свежеприготовленный раствор массовой концентрации 10 г/дм3;
- водорода пероксид по ГОСТ 10929;
- родамин С (В), раствор массовой концентрации 0,4 г/дм3 в растворе соляной кислоты молярной концентрации 1 моль/дм3;
- церий (IV) сернокислый, раствор массовой концентрации 42 г/дм3;
- изопропиловый эфир;
- сурьму трехокись;
- сурьму по ГОСТ 1089;
- растворы сурьмы известной концентрации.
14.3.2 Метод анализа
Метод основан на образовании окрашенного соединения хлоркомплекса сурьмы (V) с родамином С (В) в органической фазе после экстракции хлоркомплекса сурьмы изопропиловым эфиром. Оптическую плотность растворов соединения сурьмы с родамином С (В) измеряют при длине волны 550 нм.
14.3.3 Подготовка к выполнению анализа
14.3.3.1 При приготовлении раствора меди навеску меди по ГОСТ 859 массой 25 г растворяют в 150 см3 соляной кислоты, прибавляя порциями 100 см3 пероксида водорода. Во избежание преждевременного разложения пероксида водорода раствор постоянно охлаждают. Раствор меди переносят в мерную колбу вместимостью 500 см3 и доливают до метки соляной кислотой, разбавленной 7:3.
14.3.3.2 При приготовлении раствора сернокислого церия навеску соли 4,2 г нагревают с 5 см3 серной кислоты до появления паров серной кислоты. К раствору приливают 50 см3 воды, нагревают до растворения солей, охлаждают и переносят в мерную колбу вместимостью 100 см3, доливают до метки водой и перемешивают.
14.3.3.3 Для построения градуировочных графиков готовят растворы сурьмы известной концентрации.
При приготовлении раствора А массовой концентрации сурьмы 0,1 мг/см3 навеску 0,120 г трехокиси сурьмы нагревают с 200 см3 соляной кислоты, разбавленной 7:3, в конической колбе вместимостью 1000 см3 со шлифом и дефлегматором до полного растворения. Раствор переносят в мерную колбу вместимостью 1000 см3 и доливают до метки соляной кислотой, разбавленной 7:3.
Такой же раствор может быть приготовлен следующим образом: навеску 0,100 г тонкорастертой сурьмы помещают в коническую колбу вместимостью 250 см3 и растворяют при сильном нагревании в 20 см3 серной кислоты. Раствор выпаривают до 5 см3, охлаждают, прибавляют 200 см3 соляной кислоты, разбавленной 7:3. Раствор переносят в мерную колбу вместимостью 1000 см3, ополаскивают колбу соляной кислотой той же концентрации и присоединяют к раствору в мерной колбе. Раствор доливают до метки соляной кислотой, разбавленной 7:3, и перемешивают.
При приготовлении раствора Б массовой концентрации сурьмы 0,025 мг/см3 пипеткой отбирают 25 см3 раствора А в мерную колбу вместимостью 100 см3, доливают до метки соляной кислотой, разбавленной 7:3, и перемешивают.
При приготовлении раствора В массовой концентрации сурьмы 0,005 мг/см3 пипеткой отбирают 20 см3 раствора Б в мерную колбу вместимостью 100 см3, доливают до метки соляной кислотой, разбавленной 7:3, и перемешивают.
Растворы Б и В устойчивы в течение 8 ч.
14.3.3.4 Построение градуировочных графиков
Градуировочные графики строят соответственно значениям содержания сурьмы, указанным в таблице 13. Для этого в стаканы вместимостью от 100 до 150 см3 отбирают 0; 0,5; 1,0; 1,5; 2,0 и 2,5 см3 раствора сурьмы Б или В и объемы раствора меди, соответствующие таблице 13.
Растворы разбавляют соляной кислотой, разбавленной 7:3, до объема 15 см3, прибавляют 1 см3 раствора сернистокислого натрия, накрывают часовым стеклом и нагревают до кипения. Далее проводят анализ, как указано в 14.3.4.2.
По найденным значениям оптической плотности и соответствующим им значениям содержания сурьмы строят градуировочный график.
|
Диапазон массовой доли сурьмы, % |
Объем раствора меди, |
Объем см3 раствора сурьмы, см3 |
Масса сурьмы, мкг |
Объем изопропилового эфира, см3 |
|
Раствор В |
||||
|
От 0,0005 до 0,006 включ. |
4 |
0,5 |
2,5 |
10 |
|
4 |
1,0 |
5,0 |
10 |
|
|
4 |
1,5 |
7,5 |
10 |
|
|
4 |
2,0 |
10,0 |
10 |
|
|
4 |
2,5 |
12,5 |
10 |
|
|
Раствор Б |
||||
|
Св. 0,006 до 0,1 включ. |
2 |
0,5 |
12,5 |
50 |
|
2 |
1,0 |
25,0 |
50 |
|
|
2 |
1,5 |
37,5 |
50 |
|
|
2 |
2,0 |
50,0 |
50 |
|
|
2 |
2,5 |
62,5 |
50 |
|
14.3.4 Выполнение анализа
14.3.4.1 Общие требования к методам анализа и требования безопасности при выполнении анализов - в соответствии с разделами 3 и 4.
14.3.4.2 Навеску меди массой, указанной в таблице 14, помещают в стакан вместимостью 400 см3 и растворяют в 50 см3 соляной кислоты, разбавленной 7:3, при добавлении небольшими порциями 15 см3 пероксида водорода. Избыток пероксида водорода и выделившийся хлор удаляют кипячением раствора.
Раствор охлаждают, переносят в мерную колбу вместимостью от 100 до 250 см3 и доливают до метки соляной кислотой, разбавленной 7:3.
Отмеряют пипеткой 10 см3 раствора в стакан вместимостью от 100 до 150 см3, приливают 1 см3 раствора сернистокислого натрия и 5 см3 соляной кислоты, разбавленной 7:3, накрывают стакан часовым стеклом и нагревают до кипения. Раствор сразу же охлаждают холодной водой (ниже 20 °С) и прибавляют 4 см3 раствора сернокислого церия (IV). Через 1 мин приливают 10 капель раствора солянокислого гидроксиламина, хорошо перемешивают и переносят содержимое стакана и 50 см3 воды в делительную воронку вместимостью от 100 до 150 см3. Отмеряют бюреткой изопропиловый эфир (таблица 14) и экстрагируют сурьму в течение 1 мин.
|
Диапазон массовой доли сурьмы, % |
Масса навески меди, г |
Объем мерной колбы, см3 |
Аликвотная часть раствора, см3 |
Объем изопропилового эфира, см3 |
|
От 0,0005 до 0,0025 включ. |
5,0 |
100 |
10 |
10 |
|
Св. 0,0025 » 0,006 » |
2,0 |
100 |
10 |
10 |
|
» 0,006 » 0,025 » |
2,5 |
100 |
10 |
50 |
|
» 0,025 » 0,06 » |
2,5 |
250 |
10 |
50 |
|
» 0,06 » 0,1» |
1,0 |
200 |
10 |
50 |
Водный раствор отбрасывают, экстракт промывают 2 см3 раствора солянокислого гидроксиламина в течение 30 с. После отделения водного раствора экстракт промывают 5 см3 1 моль/дм3 раствора соляной кислоты. Водный раствор отделяют, а к органическому слою добавляют 4 см3 раствора родамина и взбалтывают содержимое воронки в течение 30 с. Избыток раствора родамина отделяют, экстракт сливают в стакан центрифуги и центрифугируют в течение 2 мин.
При отсутствии центрифуги экстракт сливают в стакан вместимостью от 100 до 150 см3, обезвоживают при помощи 0,5 г сернокислого натрия и помещают в цилиндр с притертой пробкой. Оптическую плотность экстракта измеряют при длине волны 550 нм в кювете толщиной поглощающего свет слоя 50 мм.
Раствором сравнения при измерении оптической плотности служит изопропиловый эфир.
Одновременно проводят два контрольных опыта со всеми применяемыми реактивами. Среднее значение оптической плотности контрольного опыта вычитают из значения оптической плотности анализируемого раствора.
Массу сурьмы в растворе устанавливают по градуировочному графику.
14.3.5 Обработка результатов анализа
14.3.5.1 Массовую долю сурьмы X, %, вычисляют по формуле
|
|
(23) |
где m1 - масса сурьмы, найденная по градуировочному графику, мкг;
V - объем анализируемого раствора, см3;
т - масса навески меди, г;
V1 - объем аликвотной части анализируемого раствора, см3.
14.3.5.2 За результат анализа принимают среднеарифметическое значение двух параллельных определений при условии, что абсолютная разность между ними в условиях повторяемости не превышает значений (при доверительной вероятности Р = 0,95) предела повторяемости г, приведенных в таблице 12.
Если расхождение между результатами параллельных определений превышает значение предела повторяемости, выполняют процедуры, изложенные в стандарте [2] (подпункт 5.2.2.1).
14.3.6 Контроль точности результатов анализа
Контроль точности результатов анализа - в соответствии с 3.14.
14.3.7 Оформление результатов анализа
Результаты анализа оформляют в соответствии с 3.15, значения погрешности результатов анализа А приведены в таблице 12.
14.3.8 Допускается определение массовой доли сурьмы экстракционно-фотометрическим методом с применением кристаллического фиолетового или бриллиантового зеленого.
14.4 Атомно-абсорбционный метод
14.4.1 Средства измерений, вспомогательные устройства, материалы, растворы
При выполнении анализа применяют следующие средства измерений, вспомогательные устройства:
- спектрофотометр атомно-абсорбционный с источником излучения на сурьму;
- весы лабораторные специального класса точности по ГОСТ 24104;
- пипетки не ниже 2-го класса точности по ГОСТ 29169 и ГОСТ 29227;
- колбы 2-25-2, 2-50-2 по ГОСТ 1770;
- стаканы В-1-100 ТС, В-1-400 ТС по ГОСТ 25336;
- компрессор воздушный.
При выполнении анализа применяют следующие материалы, растворы:
- ацетилен по ГОСТ 5457;
- пропан-бутан по ГОСТ 20448;
- кислоту соляную по ГОСТ 3118, раствор молярной концентрации 1 моль/дм3 и разбавленную 1:1;
- кислоту азотную по ГОСТ 4461, раствор молярной концентрации 1 моль/дм3 и разбавленную 1:1;
- кислоту серную по ГОСТ 4204;
- аммиак водный по ГОСТ 3760 и разбавленный 1:99;
- железо по ГОСТ 9849, раствор массовой концентрации 25 г/дм3 в растворе азотной кислоты молярной концентрации 1 моль/дм3;
- медь по ГОСТ 859;
- сурьму по ГОСТ 1089;
- сурьму трехокись;
- растворы сурьмы известной концентрации;
- фильтры обеззоленные по [21].
14.4.2 Метод анализа
Атомно-абсорбционный метод основан на измерении поглощения линии сурьмы в пламени ацетилен-воздух или пропан-бутан-воздух при длине волны 217,6 нм. При массовой доле сурьмы до 0,01% предварительно выделяют ее соосаждением на гидроксиде железа и растворением осадка в соляной кислоте.
14.4.3 Подготовка к выполнению анализа
14.4.3.1 Для построения градуировочных графиков готовят растворы сурьмы известной концентрации согласно 14.3.3.3.
14.4.3.2 Построение градуировочного графика
Для построения градуировочного графика в стаканы вместимостью 400 см3 каждый помещают 1,0 или 5,0 г меди (в зависимости от массовой доли сурьмы) и растворяют в соответствии с 14.4.4.2 и 14.4.4.3. Затем добавляют 0; 0,25; 0,50; 1,00; 2,00; 3,00; 5,00 и 10,0 см3 раствора А, что соответствует 0; 25; 50; 100; 200; 300; 500 и 1000 мкг сурьмы, и проводят анализ в соответствии с 14.4.4.2 или 14.4.4.3.
По полученным значениям оптической плотности и соответствующим им значениям концентрации сурьмы строят градуировочный график.
При построении графика значение сигнала фонового раствора вычитают из значения сигнала каждого раствора и проводят график из начала координат.
14.4.4 Выполнение анализа
14.4.4.1 Общие требования к методам анализа и требования безопасности при выполнении анализов - в соответствии с разделами 3 и 4.
14.4.4.2 Навеску меди массой 5,0000 г (при массовой доле сурьмы до 0,01%) помещают в стакан вместимостью 400 см3 и растворяют при нагревании в 25 см3 азотной кислоты, разбавленной 1:1. После удаления оксидов азота приливают 250 см3 воды и 5 см3 раствора железа, нагревают при температуре от 60°С до 70°С. Добавляют аммиак в таком количестве, чтобы вся медь перешла в аммиачный комплекс (синяя окраска раствора) и еще 5 см3.
Оставляют при 60°С до коагуляции осадка, после чего фильтруют на фильтр средней плотности «белая лента». Промывают осадок на фильтре 2 - 3 раза горячим раствором аммиака 1:99.
Растворяют осадок в 10 см3 соляной кислоты, разбавленной 1:1, собирая фильтрат в мерную колбу вместимостью 25 см3, и промывают фильтр 2 - 3 раза горячей водой. После охлаждения разбавляют водой до метки и перемешивают. Измеряют поглощение анализируемого раствора при длине волны 217,7 нм одновременно с раствором контрольного опыта и растворами для построения градуировочного графика.
Массу сурьмы устанавливают по градуировочному графику.
Допускается определение в анализируемом растворе висмута (от 0,0005% до 0,005%), олова (от 0,01% до 0,06%) и свинца (от 0,0005% до 0,005%).
14.4.4.3 Навеску меди массой 1,0000 г (при массовой доле сурьмы свыше 0,01%) помещают в стакан вместимостью 100 см3 и растворяют в объеме от 10 до 15 см3 азотной кислоты, разбавленной 1:1. После растворения навески приливают 10 см3 соляной кислоты, разбавленной 1:1, переводят в мерную колбу вместимостью 50 см3, разбавляют до метки раствором соляной кислоты 1 моль/дм3 и перемешивают.
Измеряют поглощение, как описано в 14.4.4.2.
14.4.5 Обработка результатов анализа
14.4.5.1 Массовую долю сурьмы X, %, вычисляют по формуле
|
|
(24) |
где т1 - масса сурьмы в растворе анализируемой пробы, найденная по градуировочному графику, мкг;
т2 - масса сурьмы в растворе контрольного опыта, мкг;
т - масса навески меди, г.
14.4.5.2 За результат анализа принимают среднеарифметическое значение двух параллельных определений при условии, что абсолютная разность между ними в условиях повторяемости не превышает значений (при доверительной вероятности Р = 0,95) предела повторяемости г, приведенных в таблице 12.
Если расхождение между результатами параллельных определений превышает значение предела повторяемости, выполняют процедуры, изложенные в стандарте [2] (подпункт 5.2.2.1).
14.4.6 Контроль точности результатов анализа
Контроль точности результатов анализа - в соответствии с 3.14.
14.4.7 Оформление результатов анализа
Результаты анализа оформляют в соответствии с 3.15, значения погрешности результатов анализа А приведены в таблице 12.
15 Методы определения массовой доли висмута
15.1 Область применения
В настоящем разделе установлены фотометрический (при массовой доле от 0,00005% до 0,02%) и атомно-абсорбционный (при массовой доле от 0,0003% до 0,005%) методы определения массовой доли висмута в меди.
15.2 Требования к погрешности анализа
Погрешность результатов анализа массовой доли висмута, значения пределов повторяемости и воспроизводимости для доверительной вероятности Р = 0,95 должны соответствовать приведенным в таблице 15.
В процентах
|
Диапазон массовой доли висмута |
Погрешность результатов анализа ±∆ |
Предел |
|
|
повторяемости r (n=2) |
воспроизводимости R |
||
|
От 0,00005 до 0,00010 включ. |
0,00001 |
0,00002 |
0,00002 |
|
Св. 0,0001 » 0,0003 » |
0,0001 |
0,0001 |
0,0002 |
|
» 0,0003 » 0,0010 » |
0,0002 |
0,0002 |
0,0003 |
|
» 0,0010 » 0,0030 » |
0,0004 |
0,0004 |
0,0006 |
|
» 0,0030 » 0,0050 » |
0,0006 |
0,0006 |
0,0009 |
|
» 0,005 » 0,020 » |
0,004 |
0,002 |
0,005 |
15.3 Фотометрический метод
15.3.1 Средства измерений, вспомогательные устройства, материалы, растворы
При выполнении анализа применяют следующие средства измерений, вспомогательные устройства:
- фотометр фотоэлектрический или спектрофотометр со всеми принадлежностями, обеспечивающие проведение измерений при длине волны от 434 до 450 нм;
- центрифугу со всеми принадлежностями;
- весы лабораторные специального класса точности по ГОСТ 24104;
- пипетки не ниже 2-го класса точности по ГОСТ 29169 и ГОСТ 29227;
- колбы мерные 2-10-2, 2-50-2, 2-500-2, 2-1000-2 по ГОСТ 1770;
- стаканы В-1-250 ТС, В-1-400 ТХС по ГОСТ 25336;
- воронку ВД-1-250 ХС по ГОСТ 25336;
- мензурку 100 по ГОСТ 1770;
- стекло часовое.
При выполнении анализа применяют следующие материалы, растворы:
- кислоту соляную по ГОСТ 3118, разбавленную 1:1 и раствор молярной концентрации 0,2 моль/дм3;
- кислоту серную по ГОСТ 4204 молярной концентрации 5 моль/дм3;
- кислоту бромистоводородную по ГОСТ 2062;
- аммиак водный по ГОСТ 3760, разбавленный 1:1;
- железо (III) хлорид 6-водный по ГОСТ 4147, раствор массовой концентрации 10 г/дм3 в растворе соляной кислоты 0,2 моль/дм3. 1 см3 раствора содержит 2 мг железа;
- натрия гидроксид (натрия гидроокись) по ГОСТ 4328, раствор массовой концентрации 80,0 г/дм3;
- хлороформ по ГОСТ 20015;
- аммоний хлористый по ГОСТ 3773, раствор массовой концентрации 200 г/дм3;
- кислоту винную по ГОСТ 5817, раствор массовой концентрации 200 г/дм3;
- гидроксиламин солянокислый по ГОСТ 5456, раствор массовой концентрации 100 г/дм3;
- калий цианистый по [25], раствор массовой концентрации 100 г/дм3;
- соль динатриевую этилендиамин-N, N, N', N'-тетрауксусной кислоты двуводную (трилон Б) по ГОСТ 10652, раствор массовой концентрации 50 г/дм3;
- натрия N, N-диэтилдитиокарбамат (Na-ДДТК) 3-водный по ГОСТ 8864, раствор;
- тимолфталеин, раствор массовой концентрации 40 г/дм3 в этиловом спирте;
- натрий углекислый кислый по ГОСТ 4201;
- промывной раствор;
- натрий фосфорноватистокислый 1-водный по ГОСТ 200, раствор массовой концентрации 300 г/дм3;
- спирт этиловый по ГОСТ 18300;
- соль закиси железа и аммония двойную сернокислую (соль Мора) по ГОСТ 4208;
- калий йодистый по ГОСТ 4232, раствор;
- висмут металлический Ви00 по ГОСТ 10928;
- растворы висмута известной концентрации;
- бумагу индикаторную универсальную по [13];
- кислоту азотную по ГОСТ 4461 и разбавленную 1:1;
- кислоту аскорбиновую, раствор массовой концентрации 50 г/дм3 свежеприготовленный;
- кислоту хлорную по [23];
- бумагу фильтровальную лабораторную по ГОСТ 12026;
- олово двухлористое по [26].
15.3.2 Метод анализа
Метод основан на образовании йодидного комплекса висмута в сернокислом растворе. Висмут отделяют от мешающих элементов соосаждением с гидроксидом железа или экстракцией диэтилдитиокарбамата висмута хлороформом. Оптическую плотность раствора йодидного комплекса висмута измеряют при длине волны от 450 до 470 нм.
15.3.3 Подготовка к выполнению анализа
15.3.3.1 При приготовлении раствора Na-ДДТК навеску 2,0 г Na-ДДТК растворяют в 100 см3 воды, в которой был предварительно растворен 1,0 г углекислого кислого натрия. Раствор фильтруют.
15.3.3.2 При приготовлении промывного раствора к 30 см3 раствора Na-ДДТК приливают 1000 см3 воды, 10 см3 раствора цианистого калия и 1 см3 раствора тимолфталеина. По каплям приливают раствор хлористого аммония до исчезновения синей окраски.
15.3.3.3 При приготовлении раствора йодистого калия навеску 200 г йодистого калия растворяют в 300 см3 воды, прибавляют 30 см3 раствора натрия фосфорноватистокислого, доливают водой до 1000 см3 и перемешивают.
15.3.3.4 Для построения градуировочных графиков готовят растворы висмута известной концентрации.
При приготовлении раствора А массовой концентрации висмута 0,1 мг/см3 навеску 0,100 г висмута растворяют в 5 см3 азотной кислоты. Оксиды азота удаляют нагреванием. Раствор охлаждают и помещают в мерную колбу вместимостью 1000 см3, приливают 65 см3 азотной кислоты, доливают водой до метки и перемешивают.
При приготовлении раствора Б массовой концентрации висмута 0,01 мг/см3 50 см3 раствора А помещают в мерную колбу вместимостью 500 см3, приливают 5 см3 азотной кислоты, доливают водой до метки и перемешивают. Полученный раствор хранят не более 5 ч.
15.3.3.5 Построение градуировочного графика
Для построения градуировочного графика в стаканы вместимостью 250 см3 каждый помещают 0; 0,5; 1,0; 2,0; 3,0; 4,0 и 5,0 см3 раствора Б, что соответствует 0; 5; 10; 20; 30; 40 и 50 мкг висмута. Во все стаканы прибавляют по 5 см3 соляной кислоты, разбавленной 1:1, 5 см3 раствора хлорида железа и доливают водой до объема от 50 до 60 см3. Затем добавляют аммиак до получения рН от 7,5 до 8,5 и 2 см3 в избытке. Раствор нагревают до температуры от 70°С до 80°С и далее поступают, как указано в 15.3.4.2 и 15.3.4.3.
По полученным значениям оптических плотностей и соответствующим значениям содержания висмута строят градуировочный график.
15.3.4 Выполнение анализа
15.3.4.1 Общие требования к методам анализа и требования безопасности при выполнении анализов - в соответствии с разделами 3 и 4.
|
Диапазон массовой доли висмута, % |
Масса навески меди, г |
|
От 0,00005 до 0,00030 включ. |
10,0000 |
|
Св. 0,0003 » 0,0010 » |
5,0000 |
|
» 0,001 » 0,005 » |
3,0000 - 1,0000 |
|
» 0,005 » 0,020 » |
0,5000 |
Раствор выпаривают примерно до 5 см3, прибавляют 100 см3 воды и 5 см3 раствора железа. Раствор нагревают до температуры от 50°С до 60°С и прибавляют раствор аммиака, разбавленный 1:1, в таком количестве, чтобы вся медь перешла в аммиачный комплекс, и еще 10 см3 (рН раствора от 7,5 до 8,5).
Содержимое стакана нагревают до температуры от 70°С до 80°С и оставляют на время от 20 до 30 мин. Осадок гидроксидов фильтруют через фильтр средней плотности и промывают на фильтре 3 - 4 раза горячей водой.
Осадок с фильтра смывают струей горячей воды в стакан, в котором проводили осаждение, и прибавляют 5 см3 соляной кислоты, разбавленной 1:1, фильтр промывают горячей водой до полного удаления железа.
К раствору приливают воду до объема 50 см3. Осаждение гидроксидов аммиаком повторяют, как указано выше. Полученный раствор выпаривают до объема от 3 до 5 см3, помещают в стакан вместимостью 100 см3 и выпаривают почти досуха. К остатку добавляют 1 см3 азотной кислоты, нагревают до появления окислов азота, охлаждают, добавляют 3 см3 хлорной кислоты и выпаривают до паров хлорной кислоты. Раствор охлаждают, добавляют 2 см3 бромистоводородной кислоты и выпаривают до влажного остатка.
К остатку прибавляют 2 см3 раствора серной кислоты, 1 см3 раствора фосфорноватистокислого натрия, 1 см3 воды и нагревают до растворения. Раствор выдерживают при температуре от 70°С до 80°С в течение 10 мин. Раствор охлаждают, помещают в мерную колбу вместимостью 10 см3, прибавляют 1 см3 раствора йодистого калия, доливают водой до метки и перемешивают. Далее поступают, как указано в 15.3.4.3.
Параллельно проводят контрольные опыты со всеми применяемыми реактивами и растворами, но без добавления меди.
15.3.4.3 Раствор выпаривают до объема 2 см3 и добавляют 10 см3 воды, 5 см3 раствора винной кислоты, 3 см3 раствора трилона Б и 0,1 см3 раствора тимолфталеина. По каплям прибавляют раствор гидроксида натрия до изменения окраски от оранжевой до синей. К раствору прибавляют 1 см3 раствора солянокислого гидроксиламина, 3 см3 раствора цианистого калия и, после перемешивания, - 3 см3 раствора хлористого аммония до рН от 9 до 10. При другом значении рН добавляют раствор гидроксида натрия или раствор хлористого аммония. Раствор переносят в делительную воронку, приливают 50 см3 воды и 1 см3 раствора трилона Б. Затем добавляют 10 см3 хлороформа и встряхивают в течение 2 мин. Органическую фазу помещают во вторую делительную воронку. Водную фазу экстрагируют дважды: сначала 10 см3, а затем 5 см3 хлороформа, встряхивая каждый раз в течение 2 мин, добавляя перед каждой экстракцией по 1 см3 раствора Na-ДДТК. Органические фазы объединяют и промывают в течение 5 мин 15 см3 промывного раствора, к которому добавляют одну каплю раствора Na-ДДТК.
Из промытой органической фазы висмут реэкстрагируют дважды, добавляя каждый раз по 10 см3 соляной кислоты, разбавленной 1:1, и встряхивая в течение 2 мин. Объединенные солянокислые реэкстракты помещают в стакан и выпаривают досуха. Остаток растворяют в 5 см3 азотной кислоты и 2 см3 хлорной кислоты и затем выпаривают досуха.
Если остаток окрашен, повторяют обработку его азотной и хлорной кислотами. Обесцвеченный остаток растворяют при нагревании в 2 см3 раствора серной кислоты. Раствор охлаждают, помещают в мерную колбу вместимостью 10 см3, прибавляют воду до объема от 6 до 7 см3 и 1 см3 раствора фосфорноватисто-кислого натрия. Через 10 мин прибавляют 1 см3 раствора йодистого калия, раствор доливают до метки водой и перемешивают.
Если раствор непрозрачный, то помещают содержимое мерной колбы в пробирку для центрифуги и центрифугируют в течение 5 с или отфильтровывают. Оптическую плотность раствора измеряют при длине волн от 450 до 470 нм в кювете толщиной поглощающего свет слоя 50 мм. Раствором сравнения служит вода.
Среднее значение оптической плотности растворов контрольных опытов вычитают из значения оптической плотности анализируемого раствора.
15.3.4.4 Определение висмута допускается проводить следующим образом: фильтрат, полученный по 15.3.4.2, нагревают до температуры от 40°С до 50°С и добавляют в него по каплям раствор двухлористого олова до потемнения раствора и еще 1 см3. Добавляют немного фильтробумажной массы, нагревают до кипения и оставляют на время от 10 до 15 мин при температуре от 70°С до 80°С до коагуляции осадка. Затем осадок фильтруют на плотный фильтр «синяя лента», в конус которого вложена фильтробумажная масса, и промывают 3 - 4 раза горячей водой. Фильтр с осадком отбрасывают.
К фильтрату приливают 4 см3 раствора винной кислоты, 5 см3 раствора йодистого калия и объем от 1,0 до 1,5 см3 раствора аскорбиновой кислоты, перемешивают, помещают полученный раствор в мерную колбу вместимостью 50 см3, разбавляют водой до метки и снова перемешивают. По истечении от 10 до 15 мин измеряют оптическую плотность при длине волны от 434 до 450 нм в кювете толщиной поглощающего свет слоя 30 мм или 50 мм. Раствором сравнения служит вода.
Массу висмута определяют по градуировочному графику.
15.3.5 Обработка результатов анализа
15.3.5.1 Массовую долю висмута X, %, вычисляют по формуле
|
|
(25) |
где m1 - масса висмута, найденная по градуировочному графику, мкг;
т - масса навески меди, г.
15.3.5.2 За результат анализа принимают среднеарифметическое значение двух параллельных определений при условии, что абсолютная разность между ними в условиях повторяемости не превышает значений (при доверительной вероятности Р = 0,95) предела повторяемости г, приведенных в таблице 15.
Если расхождение между наибольшим и наименьшим результатами параллельных определений превышает значение предела повторяемости, выполняют процедуры, изложенные в стандарте [2] (подпункт 5.2.2.1).
15.3.6 Контроль точности результатов анализа
Контроль точности результатов анализа - в соответствии с 3.14.
15.3.7 Оформление результатов анализа
Результаты анализа оформляют в соответствии с 3.15, значения погрешности результатов анализа Δ приведены в таблице 15.
15.4 Атомно-абсорбционный метод
15.4.1 Средства измерений, вспомогательные устройства, материалы, растворы
При выполнении анализа применяют следующие средства измерений, вспомогательные устройства:
- спектрофотометр атомно-абсорбционный с источником излучения на висмут;
- весы лабораторные специального класса точности по ГОСТ 24104;
- пипетки не ниже 2-го класса точности по ГОСТ 29169 и ГОСТ 29227;
- колбы мерные 2-25-2, 2-50-2, 2-1000-2 по ГОСТ 1770;
- стаканы В-1-400 ТХС по ГОСТ 25336;
- компрессор воздушный.
При выполнении анализа применяют следующие материалы, растворы:
- ацетилен по ГОСТ 5457;
- кислоту соляную по ГОСТ 3118 и разбавленную 1:1;
- кислоту азотную по ГОСТ 4461, раствор молярной концентрации 1 моль/дм3 и разбавленную 1:1;
- аммиак водный по ГОСТ 3760 и раствор 1:99;
- железо по ГОСТ 9849, раствор массовой концентрации 15 г/дм3 в растворе азотной кислоты 1 моль/дм3;
- медь по ГОСТ 859;
- висмут марки Ви00 по ГОСТ 10928;
- растворы висмута известной концентрации.
15.4.2 Метод анализа
Атомно-абсорбционный метод основан на растворении пробы в азотной кислоте, отделении висмута на гидроксиде железа и последующем измерении поглощения висмута в солянокислом растворе при длине волны 223,1 нм в пламени ацетилен-воздух.
15.4.3 Подготовка к выполнению анализа
15.4.3.1 Для построения градуировочных графиков готовят растворы висмута известной концентрации согласно 15.3.3.4.
15.4.3.2 Построение градуировочного графика
Для построения градуировочного графика в стаканы вместимостью 400 см3 каждый помещают по 5,0 г или 1,0 г меди в зависимости от массовой доли висмута и растворяют в соответствии с 15.4.4.2. Затем добавляют 0; 1,5; 5,0; 10,0; 15,0; 20,0 и 25,0 см3 раствора Б, что соответствует 0; 15; 50; 100; 150; 200 и 250 мкг висмута, или 0; 0,5; 1,0; 2,0; 3,0; 5,0 см3 раствора А, что соответствует 0; 50; 100; 200; 300 и 500 мкг висмута, и проводят анализ в соответствии с 15.3.3.5.
По полученным значениям оптической плотности и соответствующим им концентрациям висмута строят градуировочный график.
15.4.4 Выполнение анализа
15.4.4.1 Общие требования к методам анализа и требования безопасности при выполнении анализов - в соответствии с разделами 3 и 4.
15.4.4.2 Навеску меди массой 5,0000 г (при массовой доле висмута от 0,0003% до 0,005%) и 1,0000 г (при массовой доле висмута свыше 0,005%) помещают в стакан вместимостью 400 см3 и растворяют в объеме от 20 до 25 см3 азотной кислоты, разбавленной 1:1. Нагревают до удаления оксидов азота. Затем приливают 250 см3 воды и 5 см3 раствора железа, нагревают до температуры от 60°С до 70°С. Добавляют аммиак в таком количестве, чтобы вся медь перешла в аммиачный комплекс, и еще 5 см3. Выдерживают при температуре 70°С до коагуляции осадка. После этого фильтруют через фильтр средней плотности «белая лента» и промывают осадок на фильтре 2 - 4 раза горячим раствором аммиака (1:99). Осадок на фильтре растворяют в 10 см3 горячего раствора соляной кислоты, промывают 2 - 3 раза горячей водой. Раствор охлаждают, помещают в мерную колбу вместимостью 25 см3, разбавляют водой до метки и перемешивают.
Измеряют оптическую плотность раствора при длине волны 223,1 нм одновременно с раствором контрольного опыта и растворами для построения градуировочного графика.
Массу висмута устанавливают по градуировочному графику.
Допускается определение в анализируемом растворе сурьмы (от 0,0005% до 0,02%), олова (от 0,01% до 0,06%) и свинца (от 0,0005% до 0,005%).
15.4.5 Обработка результатов анализа
15.4.5.1 Массовую долю висмута X, %, вычисляют по формуле
|
|
(26) |
где m1 - масса висмута, найденная по градуировочному графику, мг;
т - масса навески меди, г.
15.4.5.2 За результат анализа принимают среднеарифметическое значение двух параллельных определений при условии, что абсолютная разность между ними в условиях повторяемости не превышает значений (при доверительной вероятности Р = 0,95) предела повторяемости г, приведенных в таблице 15.
Если расхождение между наибольшим и наименьшим результатами параллельных определений превышает значение предела повторяемости, выполняют процедуры, изложенные в стандарте [2] (подпункт 5.2.2.1).
15.4.6 Контроль точности результатов анализа
Контроль точности результатов анализа - в соответствии с 3.14.
15.4.7 Оформление результатов анализа
Результаты анализа оформляют в соответствии с 3.15, значения погрешности результатов анализа Δ приведены в таблице 15.
16 Метод определения массовой доли хрома и кадмия
16.1 Область применения
Настоящий раздел устанавливает определение массовой доли хрома в диапазоне от 0,00005% до 0,00055% и кадмия в диапазоне от 0,00002% до 0,00060% в меди атомно-абсорбционным методом.
16.2 Требования к погрешности анализа
Погрешность результатов анализа массовой доли хрома и кадмия, значения пределов повторяемости и воспроизводимости для доверительной вероятности Р = 0,95 должны соответствовать приведенным в таблице 17.
В процентах
|
Компонент |
Диапазон массовых долей компонента |
Погрешность результатов анализа ±Δ |
Предел |
|
|
повторяемости r (n=2) |
воспроизводимости R |
|||
|
Хром |
От 0,00005 до 0,00015 включ. |
0,00004 |
0,00004 |
0,00006 |
|
Св. 0,00015 » 0,00055 » |
0,00010 |
0,00010 |
0,00014 |
|
|
Кадмий |
От 0,00002 до 0,00005 включ. |
0,00002 |
0,00002 |
0,00003 |
|
Св. 0,00005 » 0,00015 » |
0,00003 |
0,00003 |
0,00004 |
|
|
» 0,00015 » 0,00045 » |
0,00006 |
0,00007 |
0,00009 |
|
|
» 0,00045 » 0,00060 » |
0,00018 |
0,00020 |
0,00025 |
|
16.3 Определение массовой доли хрома
16.3.1 Средства измерений, вспомогательные устройства, материалы, растворы
При выполнении анализа применяют следующие средства измерений, вспомогательные устройства:
- электролизную установку постоянного тока;
- электроды из платины сетчатые по ГОСТ 6563;
- спектрофотометр атомно-абсорбционный с источником излучения на хром;
- компрессор воздушный;
- весы лабораторные специального класса точности по ГОСТ 24104;
- колбы мерные 2-100-2, 2-1000-2 по ГОСТ 1770;
- пробирки П-2-10-0,2 ХС по ГОСТ 1770;
- цилиндры 2-100-2 по ГОСТ 1770;
- стаканы Н-1- 50, Н-1-100, Н-1- 250, Н-1-400 ТС по ГОСТ 25336;
- пипетки 1-2-1-5 и 1-2-1-10 по ГОСТ 29227;
- стекло часовое.
При выполнении анализа применяют следующие материалы, растворы:
- ацетилен по ГОСТ 5457;
- кислоту азотную особой чистоты по ГОСТ 11125 или кислоту азотную по ГОСТ 4461, прокипяченную до удаления оксидов азота или перегнанную в кварцевом аппарате, разбавленную 1:1;
- кислоту соляную особой чистоты по ГОСТ 14261 или кислоту соляную по ГОСТ 3118, разбавленную 1:1;
- кислоту серную особой чистоты по ГОСТ 14262 или кислоту серную по ГОСТ 4204, разбавленную 1:1;
- калий двухромовокислый по ГОСТ 4220, перекристаллизованный и высушенный до постоянной массы при температуре от 140°С до 150°С;
- хром по ГОСТ 5905;
- медь по ГОСТ 859, раствор массовой концентрации 20 г/дм3;
- калий хромовокислый по ГОСТ 4459;
- воду дистиллированную по ГОСТ 6709, дополнительно очищенную по ГОСТ 4517 перегонкой в кварцевом аппарате (или стеклянном приборе);
- водорода пероксид по ГОСТ 10929;
- СО состава меди с аттестованной характеристикой хрома (кадмия).
16.3.2 Метод анализа
Метод основан на измерении поглощения резонансной линии хрома при длине волны 357,9 нм при введении анализируемого раствора в пламя ацетилен-воздух после отделения меди электролизом.
16.3.3 Подготовка к выполнению анализа
16.3.3.1 Приготовление растворов хрома известной концентрации
Раствор А
Метод 1: навеску двухромовокислого калия массой 0,2828 г, предварительно перекристаллизованного и высушенного, помещают в стакан вместимостью 100 см3, приливают 20 см3 воды и перемешивают до растворения навески. Затем добавляют 1 см3 серной кислоты и, после охлаждения, приливают пероксид водорода до прекращения бурного выделения газа. Оставляют при комнатной температуре до полного исчезновения желтой окраски (несколько часов). Затем переводят в мерную колбу вместимостью 1000 см3, приливают 10 см3 соляной кислоты, разбавляют водой до метки и перемешивают.
1 см3 раствора А содержит 0,1 мг хрома.
Метод 2: навеску хрома массой 0,1000 г помещают в стакан вместимостью 100 см3, добавляют 10 см3 соляной кислоты и нагревают до растворения навески на кипящей водяной бане. Затем раствор упаривают до сухих солей, добавляют 5 см3 азотной кислоты, и раствор выпаривают до влажных солей. Операцию обработки солей азотной кислотой проводят еще три раза. К влажным солям в стакане приливают от 50 до 70 см3 азотной кислоты, разбавленной 1:1, помещают полученный раствор в мерную колбу вместимостью 1000 см3, доливают до метки азотной кислотой, разбавленной 1:1, и перемешивают.
От полученного раствора отбирают аликвотную часть от 10 до 20 см3 и помещают ее в стакан вместимостью 50 см3 для проверки на присутствие в растворе хлорид-иона с использованием азотнокислого серебра. Если в растворе обнаружен хлорид-ион, обработку пробы хрома азотной кислотой повторяют.
Метод 3: навеску хромовокислого калия массой 0,3735 г помещают в мерную колбу вместимостью 1000 см3, растворяют в воде, доводят до метки водой и перемешивают. Раствор готовят перед применением, его свойства сохраняются в течение одних суток.
Раствор Б
Аликвотную часть (10 см3) раствора А помещают в мерную колбу вместимостью 100 см3, разбавляют водой до метки и перемешивают.
1 см3 раствора Б содержит 0,01 мг хрома.
16.3.3.2 Приготовление растворов сравнения
В мерные колбы вместимостью 100 см3 каждая помещают 0; 1,0; 2,0; 5,0 и 10 см3 раствора Б, что соответствует 0; 0,1; 0,2; 0,5 и 1,0 мг/дм3 хрома, доводят водой до метки и перемешивают.
16.3.3.3 Построение градуировочного графика
Растворы сравнения распыляют в пламени ацетилен-воздух, регистрируя поглощение при длине волны 357,9 нм для всех градуировочных растворов, последовательно от меньшей массовой концентрации к большей и в обратном порядке.
По полученным значениям оптических плотностей растворов и соответствующим им значениям массовых концентраций строят градуировочный графике прямоугольных координатах. При построении графика для каждой точки используют среднее значение двух измерений оптической плотности.
Для построения градуировочного графика готовят растворы кадмия известной концентрации. Для этого в мерные колбы вместимостью 100 см3 каждая помещают 0; 0,2; 0,4; 1,0; 2,0; 4,0; 10,0 см3 раствора Б и 1,0 см3 раствора А, приготовленных по 16.4.3.2, что соответствует 0; 0,01; 0,02; 0,05; 0,1; 0,2; 0,5 и 1,0 мг (0,0; 0,1; 0,2; 0,5; 1,0; 2,0; 5,0; 10,0 мг/дм3) кадмия. Доливают водой до метки, перемешивают. Растворы распыляют в пламени ацетилен-воздух и измеряют поглощение линии кадмия при длине волны 228,8 нм последовательно по возрастанию массовой концентрации элемента в растворе и в обратном порядке. По полученным данным строят градуировочный график в прямоугольных координатах.
16.3.4 Выполнение анализа
16.3.4.1 Общие требования к методам анализа, средствам измерений и требования безопасности при выполнении измерений - в соответствии с разделами 3 и 4.
16.3.4.2 Массовую долю хрома и кадмия определяют не менее чем по двум навескам меди. Одновременно с проведением анализа в тех же условиях проводят два контрольных опыта для внесения соответствующей поправки в результат анализа, вычитая значение контрольного опыта из результата определения при анализе пробы.
16.3.4.3 Масса навески меди и вместимость мерной посуды в зависимости от массовой доли хрома приведены в таблице 18.
|
Массовая доля хрома, % |
Масса навески, г |
Вместимость мерной посуды, см3 |
Масса хрома в растворе сравнения, мг |
|
От 0,00005 до 0,00015 включ. |
4,00 |
10,0 |
0,4 |
|
Св. 0,00015 » 0,00055 » |
2,00 |
10,0 |
1,0 |
16.3.4.4 Навеску меди помещают в стакан вместимостью 250 см3, приливают от 20 до 30 см3 азотной кислоты, разбавленной 1:1, накрывают часовым стеклом и выдерживают без нагревания до прекращения выделения оксидов азота. Затем стекло снимают, обмывают его водой над стаканом, раствор в стакане нагревают до растворения навески.
Приливают воды до объема 200 см3 и добавляют 5 см3 серной кислоты, разбавленной 1:1. В полученный раствор опускают платиновые сетчатые электроды и подвергают электролизу при плотности тока от 2,0 до 3,0 А/дм3 и напряжении от 2,2 до 2,5 В при перемешивании. Когда раствор обесцветится, электролиз прекращают. Электроды вынимают и промывают водой, а электролит упаривают до сухих солей. Затем соли растворяют в объеме от 3 до 5 см3 соляной кислоты, разбавленной 1:1, и раствор помещают в мерную пробирку вместимостью 10 см3, доводят до метки водой и перемешивают.
Полученный раствор, растворы контрольного опыта и растворы сравнения распыляют в обогащенном воздушно-ацетиленовом пламени, регистрируя поглощение при длине волны 357,9 нм. Условия измерения подбирают в соответствии с маркой (моделью) используемого прибора.
Для градуировки прибора применяют растворы сравнения массовой концентрации хрома от 0,1 до 1,0 мг/дм3.
16.3.4.5 После измерения массовой концентрации хрома в анализируемом растворе допускается проводить определение массовой концентрации кадмия, распыляя раствор в пламени ацетилен-воздух и измеряя поглощение резонансной линии кадмия при длине волны 228,8 нм. Массу кадмия определяют по градуировочному графику, построенному, как указано в 16.3.3.4.
16.3.5 Обработка результатов анализа
16.3.5.1 Массовую долю хрома X, %, вычисляют по формуле
|
|
(27) |
где m1 - массовая концентрация хрома в анализируемом растворе, найденная по градуировочному графику, мг/дм3;
m2 - концентрация хрома в растворе контрольного опыта, мг/дм3;
V - вместимость мерной пробирки, см3;
т - масса навески меди, г.
Аналогично рассчитывают массовую долю кадмия.
16.3.5.2 За результат анализа принимают среднеарифметическое значение двух параллельных определений при условии, что абсолютная разность между ними в условиях повторяемости не превышает значений (при доверительной вероятности Р = 0,95) пределов повторяемости г, приведенных в таблице 17. Если расхождение между результатами параллельных определений превышает значение предела повторяемости, выполняют процедуры, изложенные в стандарте [2] (подпункт 5.2.2.1).
16.3.6 Контроль точности результатов анализа
Контроль точности результатов анализа - в соответствии с 3.14.
16.3.7 Оформление результатов анализа
Результаты анализа оформляют в соответствии с 3.15, значения погрешности результатов анализа А приведены в таблице 17.
16.4 Определение массовой доли кадмия
16.4.1 Средства измерений, вспомогательные устройства, материалы, растворы
При выполнении анализа применяют следующие средства измерений, вспомогательные устройства:
- спектрофотометр атомно-абсорбционный с источником излучения на хром;
- компрессор воздушный;
- весы лабораторные специального класса точности по ГОСТ 24104;
- стаканы Н-1-100 ТХС по ГОСТ 25336;
- колбы мерные 2-100-2, 2-1000-2 по ГОСТ 1770;
- цилиндр 2-100-1 по ГОСТ 1770;
- пипетки не ниже 2-го класса точности по ГОСТ 29169 и ГОСТ 29227.
При выполнении анализа применяют следующие материалы, растворы:
- ацетилен по ГОСТ 5457;
- кислоту азотную особой чистоты по ГОСТ 11125 или кислоту азотную по ГОСТ 4461, разбавленную 1:1;
- медь по ГОСТ 859, раствор массовой концентрации 20 г/дм3;
- кадмий по ГОСТ 1467, марка КдО.
16.4.2 Метод анализа
Метод основан на измерении поглощения резонансной линии кадмия при длине волны 228,8 нм при введении анализируемого раствора в воздушно-ацетиленовое пламя.
16.4.3 Подготовка к выполнению анализа
16.4.3.1 При приготовлении раствора меди массовой концентрации 20 г/дм3 навеску меди массой 2,00 г растворяют при нагревании в объеме от 15 до 20 см3 азотной кислоты до удаления оксидов азота. Охлаждают и помещают раствор в мерную колбу вместимостью 100 см3, доливают до метки водой и перемешивают.
16.4.3.2 Приготовление растворов кадмия известной концентрации
Раствор А: навеску кадмия массой 1,0000 г растворяют в 15 см3 азотной кислоты и 10 см3 воды при нагревании до растворения навески и прекращения выделения паров оксидов азота. Затем раствор помещают в мерную колбу вместимостью 1000 см3, разбавляют водой до метки и перемешивают.
1 см3 раствора А содержит 1 мг кадмия.
Раствор Б: отбирают пипеткой 5 см3 раствора А и помещают в мерную колбу вместимостью 100 см3, разбавляют водой до метки и перемешивают.
1 см3 раствора Б содержит 0,05 мг кадмия.
Раствор В: отбирают пипеткой 10 см3 раствора Б и помещают в мерную колбу вместимостью 100 см3, разбавляют водой до метки и перемешивают.
1 см3 раствора В содержит 0,005 мг кадмия.
16.4.3.3 Построение градуировочного графика
В мерные колбы вместимостью 100 см3 каждая помещают: 0; 0,5; 1,0; 3,0; 5,0; 10 см3 раствора В, что соответствует 0; 0,0025; 0,005; 0,015; 0,025; 0,050 мг кадмия. Приливают по 50 см3 раствора меди, разбавляют водой до метки и перемешивают. Градуировочные растворы распыляют в пламени ацетилен-воздух, регистрируя поглощение линии кадмия при длине волны 228,8 нм последовательно от меньшей к большей массовой концентрации и в обратном порядке.
По полученным значениям оптических плотностей градуировочных растворов и соответствующим им значениям массовых концентраций кадмия строят график в прямоугольных координатах. При построении графика для каждой точки используют среднее значение двух измерений оптической плотности.
16.4.4 Выполнение анализа
16.4.4.1 Общие требования к методам анализа, средствам измерений и требования безопасности при выполнении измерений - в соответствии с разделами 3 и 4.
16.4.4.2 Массовую долю кадмия определяют не менее чем по двум навескам меди. Одновременно с проведением анализа в тех же условиях проводят контрольный опыт для внесения соответствующей поправки в результат анализа, вычитая значение контрольного опыта из результата определения при анализе пробы.
16.4.4.3 Навеску меди массой 1,0000 г помещают в стакан вместимостью 100 см3, приливают от 10 до 15 см3 азотной кислоты и оставляют без нагревания до прекращения выделения паров оксидов азота. Затем раствор нагревают до полного растворения навески, добавляют от 30 до 50 см3 воды и помещают полученный раствор в мерную колбу вместимостью 100 см3, разбавляют водой до метки и перемешивают. Раствор распыляют в пламени ацетилен-воздух и измеряют поглощение линии кадмия при длине волны 228,8 нм.
Массу кадмия определяют по градуировочному графику.
Условия измерения подбирают в соответствии с используемым прибором.
16.4.5 Обработка результатов анализа
16.4.5.1 Массовую долю кадмия X, %, вычисляют по формуле
|
|
(28) |
где m1 - масса кадмия в пробе, найденная по градуировочному графику, мг;
т - масса навески меди, г.
16.4.5.2 За результат анализа принимают среднеарифметическое значение двух параллельных определений при условии, что абсолютная разность между ними в условиях повторяемости не превышает значений (при доверительной вероятности Р = 0,95) предела повторяемости г, приведенных в таблице 17.
Если расхождение между результатами параллельных определений превышает значение предела повторяемости, выполняют процедуры, изложенные в стандарте [2] (подпункт 5.2.2.1).
16.4.6 Контроль точности результатов анализа
Контроль точности результатов анализа - в соответствии с 3.14.
16.4.7 Оформление результатов анализа
Результаты анализа оформляют в соответствии с 3.15, значения погрешности результатов анализа А приведены в таблице 17.
17 Метод спектрального анализа с фотоэлектрической регистрацией спектра
17.1 Область применения
В настоящем разделе установлено определение массовых долей примесей в меди по ГОСТ 859 в диапазонах, приведенных в таблице 19, методом атомно-эмиссионной спектрометрии с фотоэлектрической регистрацией спектра.
В процентах
|
Определяемый элемент |
Диапазон массовых долей элемента |
Определяемый элемент |
Диапазон массовых долей элемента |
|
Висмут |
От 0,00006 до 0,015 включ. |
Сурьма |
От 0,00015 до 0,090 включ. |
|
Селен |
От 0,00008 до 0,00030 включ. |
Мышьяк |
От 0,00005 до 0,030 включ. |
|
Теллур |
От 0,00010 до 0,00030 включ. |
Марганец |
От 0,00005 до 0,0030 включ. |
|
Никель |
От 0,00010 до 0,30 включ. |
Железо |
От 0,00010 до 0,080 включ. |
|
Цинк |
От 0,00010 до 0,0060 включ. |
Свинец |
От 0,00015 до 0,060 включ. |
|
Кремний |
От 0,00004 до 0,0010 включ. |
Сера |
От 0,0005 до 0,030 включ. |
|
Олово |
От 0,00008 до 0,090 включ. |
Серебро |
От 0,0006 до 0,0060 включ. |
|
Кобальт |
От 0,00005 до 0,00020 включ. |
Кадмий |
От 0,00004 до 0,00015 включ. |
|
Фосфор |
От 0,00004 до 0,090 включ. |
Хром |
От 0,00004 до 0,00060 включ. |
17.2 Требования к погрешности измерений
Погрешности результатов измерений массовых долей элементов, значения пределов повторяемости и воспроизводимости для доверительной вероятности Р = 0,95 должны соответствовать приведенным в таблице 20.
В процентах
|
Наименование элемента |
Диапазон массовых долей элемента |
Погрешность результатов измерений ±Δ |
Предел |
|
|
Повторяемости r (n=2) |
воспроизводимости R |
|||
|
Висмут |
От 0,00006 до 0,00030 включ. |
0,00006 |
0,00005 |
0,00008 |
|
Св. 0,00030 » 0,00050 » |
0,00017 |
0,00017 |
0,00023 |
|
|
» 0,0005 » 0,0015 » |
0,0004 |
0,0003 |
0,0006 |
|
|
» 0,0015 » 0,0050 » |
0,0009 |
0,0009 |
0,0013 |
|
|
» 0,005 » 0,015 » |
0,002 |
0,002 |
0,003 |
|
|
Селен |
От 0,00008 до 0,00030 включ. |
0,00007 |
0,00005 |
0,00009 |
|
Теллур |
От 0,00010 до 0,00030 включ. |
0,00006 |
0,00005 |
0,00008 |
|
Никель |
От 0,00010 до 0,00030 включ. |
0,00006 |
0,00006 |
0,00008 |
|
Св. 0,00030 » 0,00100 включ. |
0,00018 |
0,00019 |
0,00025 |
|
|
» 0,0010 » 0,0030 » |
0,0006 |
0,0006 |
0,0008 |
|
|
» 0,003 » 0,010 » |
0,002 |
0,002 |
0,003 |
|
|
» 0,010 » 0,030 » |
0,004 |
0,004 |
0,006 |
|
|
» 0,030 » 0,100 » |
0,014 |
0,010 |
0,024 |
|
|
» 0,10 » 0,30 » |
0,04 |
0,03 |
0,06 |
|
|
Цинк |
От 0,00010 до 0,00020 включ. |
0,00009 |
0,00010 |
0,00012 |
|
Св. 0,00020 » 0,00060 » |
0,00015 |
0,00009 |
0,00020 |
|
|
» 0,0006 » 0,0020 » |
0,0002 |
0,0002 |
0,0004 |
|
|
» 0,0020 » 0,0060 » |
0,0005 |
0,0004 |
0,0006 |
|
|
Кремний |
От 0,00004 до 0,00015 включ. |
0,00004 |
0,00003 |
0,00005 |
|
Св. 0,00015 » 0,00050 » |
0,00006 |
0,00006 |
0,00009 |
|
|
» 0,0005 » 0,0010 » |
0,0004 |
0,0004 |
0,0005 |
|
|
Олово |
От 0,00008 до 0,00030 включ. |
0,00006 |
0,00005 |
0,00007 |
|
Св. 0,00030 » 0,00100 » |
0,00014 |
0,00014 |
0,00020 |
|
|
» 0,0010 » 0,0030 » |
0,0005 |
0,0004 |
0,0006 |
|
|
» 0,0030 » 0,0090 » |
0,0013 |
0,0013 |
0,0019 |
|
|
» 0,009 » 0,030 » |
0,004 |
0,003 |
0,005 |
|
|
» 0,030 » 0,090 » |
0,010 |
0,009 |
0,015 |
|
|
Марганец |
От 0,00005 до 0,00020 включ. |
0,00005 |
0,00004 |
0,00008 |
|
Св. 0,0002 » 0,0010 » |
0,0001 |
0,0001 |
0,0002 |
|
|
» 0,0010 » 0,0030 » |
0,0007 |
0,0006 |
0,0010 |
|
|
Сурьма |
От 0,00015 до 0,00060 включ. |
0,00009 |
0,00009 |
0,00015 |
|
Св. 0,0006 » 0,0020 » |
0,0004 |
0,0004 |
0,0006 |
|
|
» 0,0020 » 0,0100 » |
0,0015 |
0,0012 |
0,0020 |
|
|
» 0,010 » 0,030 » |
0,005 |
0,004 |
0,008 |
|
|
» 0,030 » 0,090 » |
0,012 |
0,012 |
0,018 |
|
|
Мышьяк |
От 0,00005 до 0,00020 включ. |
0,00004 |
0,00004 |
0,00006 |
|
Св. 0,0002 » 0,0006 » |
0,0001 |
0,0001 |
0,0002 |
|
|
» 0,0006 » 0,0010 » |
0,0004 |
0,0003 |
0,0005 |
|
|
» 0,0010 » 0,0030 » |
0,0008 |
0,0007 |
0,0008 |
|
|
» 0,003 » 0,010 » |
0,002 |
0,002 |
0,003 |
|
|
» 0,010 » 0,030 » |
0,004 |
0,004 |
0,007 |
|
|
Хром |
От 0,00004 до 0,00010 включ. |
0,00003 |
0,00003 |
0,00005 |
|
Св. 0,00010 » 0,00060 » |
0,00007 |
0,00007 |
0,00009 |
|
|
Фосфор |
От 0,00004 до 0,00015 включ. |
0,00004 |
0,00004 |
0,00006 |
|
Св. 0,00015 » 0,00060 » |
0,00011 |
0,00011 |
0,00015 |
|
|
» 0,0006 » 0,0010 » |
0,0004 |
0,0003 |
0,0005 |
|
|
» 0,0010 » 0,0030 » |
0,0008 |
0,0007 |
0,0010 |
|
|
» 0,0030 » 0,0100 » |
0,0014 |
0,0010 |
0,0020 |
|
|
» 0,010 » 0,030 » |
0,004 |
0,003 |
0,006 |
|
|
» 0,030 » 0,090 » |
0,011 |
0,011 |
0,015 |
|
|
Железо |
От 0,00010 до 0,00040 включ. |
0,00009 |
0,00008 |
0,00011 |
|
Св. 0,0004 » 0,0020 » |
0,0003 |
0,0003 |
0,0004 |
|
|
» 0,0020 » 0,0060 » |
0,0009 |
0,0007 |
0,0011 |
|
|
» 0,006 » 0,030 » |
0,004 |
0,003 |
0,005 |
|
|
» 0,030 » 0,080 » |
0,014 |
0,014 |
0,019 |
|
|
Свинец |
От 0,00015 до 0,00060 включ. |
0,00010 |
0,00010 |
0,00015 |
|
Св. 0,0006 » 0,0020 » |
0,0004 |
0,0004 |
0,0005 |
|
|
» 0,0020 » 0,0060 » |
0,0013 |
0,0013 |
0,0019 |
|
|
» 0,006 » 0,010 » |
0,003 |
0,003 |
0,004 |
|
|
» 0,010 » 0,030 » |
0,007 |
0,007 |
0,009 |
|
|
» 0,030 » 0,060 » |
0,011 |
0,011 |
0,016 |
|
|
Кадмий |
От 0,00004 до 0,00015 включ. |
0,00003 |
0,00004 |
0,00007 |
|
Кобальт |
От 0,00005 до 0,00020 включ. |
0,00005 |
0,00004 |
0,00007 |
|
Серебро |
От 0,0006 до 0,0020 включ. |
0,0002 |
0,0002 |
0,0003 |
|
Св. 0,0020 » 0,0060 » |
0,0004 |
0,0003 |
0,0004 |
|
|
Сера |
От 0,0005 до 0,0015 включ. |
0,0002 |
0,0002 |
0,0003 |
|
Св. 0,0015 » 0,0030 » |
0,0004 |
0,0004 |
0,0006 |
|
|
» 0,0030 » 0,0100 » |
0,0010 |
0,0008 |
0,0012 |
|
|
» 0,010 » 0,030 » |
0,003 |
0,002 |
0,004 |
|
17.3 Средства измерений, вспомогательные устройства, материалы, растворы
При выполнении измерений применяют следующие средства измерений, вспомогательные устройства:
- атомно-эмиссионный спектрометр «SPECTROLAB S» или аналогичный;
- станок для заточки поверхностных проб;
- компрессор модели MI-7 или аналогичный;
- манометр диапазоном измерений от 0 до 10 МПа;
- печь очистки аргона «Rare Gas Purifier MP-2000».
При выполнении измерений применяют следующие материалы:
- СО состава меди ГСО 7284-96 (комплект СОМ) или другие близкие по составу;
- аргон по ГОСТ 10157;
- воздух сжатый под давлением от 4 до 6 МПа.
17.4 Метод измерений
Метод основан на измерении интенсивности спектральных линий определяемых элементов в анализируемом образце монолитной формы и образцах сравнения с использованием атомно-эмиссионного спектрометра «SPECTROLAB S» или другого типа с дуговым и искровым источником возбуждения и фотоэлектрической регистрацией спектра.
17.5 Подготовка к выполнению измерений
17.5.1 Подготовка прибора
Подготовку прибора к выполнению измерений проводят в соответствии с требованиями действующей инструкции по эксплуатации спектрометра «SPECTROLAB S».
Спектрометр градуируют при создании метода с использованием СО состава меди и строят зависимость интенсивности аналитической линии от массовой доли для каждого определяемого элемента. При дальнейшей работе выполняют корректировку градуировочных характеристик в соответствии с инструкцией по эксплуатации спектрометра «SPECTROLAB S».
17.5.2 Требования к пробам
Пробы на измерение должны поступать в виде:
- монолитного образца (темплета), имеющего хотя бы одну плоскую поверхность диаметром не менее 20 мм и высотой не менее 10 мм;
- стержней.
При проведении измерений стержневых образцов используют адаптер или проводят прессование стержня для получения плоской поверхности.
17.5.3 Подготовка проб к измерению
Поверхность пробы непосредственно перед проведением измерения обрабатывают на фрезерном станке и другом оборудовании, обеспечивающем получение ровной поверхности без раковин, царапин, трещин и шлаковых включений. Обработку осуществляют в соответствии с рабочей инструкцией по эксплуатации оборудования.
17.6 Выполнение измерений
17.6.1 Общие требования к методам измерений и требования безопасности при выполнении измерений - в соответствии с разделами 3 и 4, отбор проб для анализа - по ГОСТ 546, ГОСТ 4960, ГОСТ 24231 и другим нормативным документам на конкретную продукцию.
17.6.2 Выполнение измерений осуществляют в соответствии с инструкцией по эксплуатации прибора. Массовую долю примесей определяют параллельно в двух образцах. Для каждого образца в виде темплета или спрессованного стержня выполняют по три измерения. Для каждого образца в виде стержня выполняют по два единичных измерения с одного конца стержня с промежуточным фрезерованием поверхности образца между измерениями, и третье измерение выполняют с другого конца стержня.
17.7 Обработка результатов измерений
Обработку результатов измерений проводит компьютер спектрометра автоматически по заданной программе и представляет их в виде значений массовых долей определяемых элементов. За результат измерений принимают среднеарифметическое значение результатов двух параллельных определений при условии, что расхождение между их значениями при доверительной вероятности Р = 0,95 не превышает значений предела повторяемости r, приведенных в таблице 20.
Если расхождение между результатами параллельных определений превышает значение предела повторяемости, выполняют процедуры, изложенные в стандарте [2] (подпункт 5.2.2.1).
17.8 Контроль точности результатов измерений
Контроль точности результатов измерений - в соответствии с 3.14.
17.9 Оформление результатов измерений
Результаты измерений оформляют в соответствии с 3.15, значения погрешности результатов измерений ∆ приведены в таблице 20.
18 Методы атомно-абсорбционного анализа
18.1 Область применения
Настоящий раздел устанавливает определение массовых долей примесей в меди в диапазонах, указанных в таблице 21, атомно-абсорбционными методами.
В процентах
|
Определяемый элемент |
Массовая доля |
|
Висмут |
0,00001 - 0,005 |
|
Железо |
0,0002 - 0,005 |
|
Марганец |
0,0002 - 0,005 |
|
Свинец |
0,0002 - 0,005 |
|
Селен |
0,00002 - 0,0005 |
|
Серебро |
0,0002 - 0,005 |
|
Сурьма |
0,0003- 0,005 |
|
Теллур |
0,00001 - 0,0002 |
|
Олово |
0,00001 - 0,0005 |
18.2 Общие требования
18.2.1 Общие требования к методам анализа, требования безопасности при выполнении анализа - в соответствии с разделами 3 и 4.
18.2.2 Массовую долю примесей определяют не менее чем по двум навескам меди.
18.2.3 Допускается последовательное определение нескольких элементов из одной навески после соответствующего разбавления и отбора аликвотных частей.
18.3 Определение массовой доли серебра
18.3.1 Требования к погрешности результатов анализа
Погрешность результатов анализа массовой доли серебра, значения пределов повторяемости и воспроизводимости для доверительной вероятности Р = 0,95 должны соответствовать приведенным в таблице 22.
В процентах
|
Диапазон массовой доли серебра |
Погрешность результатов анализа ±∆ |
Предел |
|
|
повторяемости r (n-2) |
воспроизводимости R |
||
|
От 0,0005 до 0,0020 включ. |
0,0001 |
0,0001 |
0,0002 |
|
Св. 0,0020 » 0,0100 » |
0,0004 |
0,0003 |
0,0005 |
18.3.2 Средства измерений, вспомогательные устройства, материалы, растворы
При выполнении анализа применяют следующие средства измерений, вспомогательные устройства:
- спектрофотометр атомно-абсорбционный с источником излучения на серебро;
- компрессор воздушный;
- весы лабораторные специального класса точности по ГОСТ 24104;
- стаканы Н-1-100 ТХС, В-1-400 ТХС по ГОСТ 25336;
- колбы мерные 2-100-2, 2-1000-2 по ГОСТ 1770;
- пипетки 5-2-10 по ГОСТ 29227;
- цилиндр 1-10 по ГОСТ 1770;
- стекло часовое.
При выполнении анализа применяют следующие материалы, растворы:
- ацетилен по ГОСТ 5457;
- пропан-бутан по ГОСТ 20448;
- фильтры по [21] или другие средней плотности;
- кислоту азотную особой чистоты по ГОСТ 11125 или кислоту азотную по ГОСТ 4461 (не содержащую хлора), разбавленную 1:1, и раствор молярной концентрации 1 моль/дм3;
- кислоту соляную по ГОСТ 3118 и растворы молярной концентрации 1; 2; 6 моль/дм3;
- аммоний роданистый по ГОСТ 27068, раствор массовой концентрации 20 г/дм3;
- серебро по ГОСТ 6836;
- смолу АВ-17 по ГОСТ 20301;
- медь по ГОСТ 859, раствор массовой концентрации 100 г/дм3.
18.3.3 Метод анализа
Метод основан на измерении при длине волны 328,1 нм резонансного излучения атомами серебра, образующимися в результате атомизации при введении анализируемого раствора в пламя ацетилен-воздух или пропан-бутан-воздух.
18.3.4 Подготовка к выполнению анализа
18.3.4.1 Подготовка смолы АВ-17
Навеску смолы массой 50 г помещают в стакан вместимостью 400 см3, заливают водой и оставляют на сутки. Затем воду сливают и смолу несколько раз заливают раствором соляной кислоты молярной концентрации 1 моль/дм3, каждый раз сливая раствор декантацией. Последняя порция слива должна показывать отрицательную реакцию на наличие железа (с раствором роданида аммония). Затем смолу заливают раствором соляной кислоты молярной концентрации 1 моль/дм3 и хранят до применения.
18.3.4.2 Очистка меди от серебра
К раствору меди добавляют от 20 до 30 г подготовленного анионита АВ-17 и встряхивают в течение 15 мин. Раствор фильтруют через неплотный фильтр, собирая фильтрат в мерную колбу вместимостью 100 см3. Остаток на фильтре промывают раствором соляной кислоты молярной концентрации 1 моль/дм3, собирая промывные воды в ту же колбу, затем доводят до метки раствором соляной кислоты молярной концентрации 1 моль/дм3. Полученный раствор используют в качестве контрольного опыта при определении серебра в меди.
18.3.4.3 Приготовление раствора меди массовой концентрации 100 г/дм3
При приготовлении раствора меди массовой концентрации 100 г/дм3 навеску меди по ГОСТ 859 массой 10 г, предварительно очищенную от серебра на анионите АВ-17 в хлоридной форме по 18.3.4.2, растворяют при нагревании в 20 см3 азотной кислоты, разбавленной 1:1, раствор упаривают до сухих солей. Затем приливают 10 см3 соляной кислоты и еще раз упаривают до сухих солей, охлаждают, прибавляют 16 см3 соляной кислоты, перемешивают, помещают полученный раствор в мерную колбу вместимостью 100 см3 и доводят водой до метки.
18.3.4.4 Приготовление растворов известной концентрации
Раствор А: навеску серебра массой 0,1000 г растворяют при нагревании в 10 см3 азотной кислоты, разбавленной 1:1. К раствору добавляют 25 см3 воды, от 100 до 20 см3 соляной кислоты, перемешивают, помещают в мерную колбу вместимостью 1000 см3 и доводят до метки раствором соляной кислоты молярной концентрации 6 моль/дм3.
1 см3 раствора А содержит 0,1 мг серебра.
Раствор Б: аликвотную часть 10 см3 раствора А помещают в мерную колбу вместимостью 100 см3 и доводят до метки раствором соляной кислоты молярной концентрации 2 моль/дм3.
1 см3 раствора Б содержит 0,01 мг серебра.
Раствор В: аликвотную часть 5 см3 раствора Б помещают в мерную колбу вместимостью 100 см3 и доводят до метки раствором соляной кислоты молярной концентрации 2 моль/дм3. Используется только свежеприготовленный раствор.
1 см3 раствора В содержит 0,0005 мг серебра.
18.3.4.5 Построение градуировочного графика
В мерные колбы вместимостью 100 см3 каждая помещают 0; 1,0; 2,0; 3,0; 5,0 и 10,0 см3 раствора В, что соответствует 0; 0,0005; 0,0010; 0,0015; 0,0025 и 0,0050 мг серебра. Во все мерные колбы приливают по 10 см3 раствора меди, по 16 см3 соляной кислоты и доводят водой до метки.
Полученные растворы распыляют в пламени ацетилен-воздух (или пропан-бутан-воздух) и измеряют поглощение линии серебра при длине волны 328,1 нм. По полученным значениям оптических плотностей растворов и соответствующим им значениям массовых долей серебра строят градуировочный график в прямоугольных координатах.
18.3.5 Выполнение анализа
Навеску меди массой 1,0000 г помещают в стакан вместимостью 100 см3, приливают 10 см3 азотной кислоты, накрывают часовым стеклом и оставляют без нагревания до прекращения выделения паров оксидов азота. Затем нагревают до растворения навески. Стекло снимают, обмывают его водой над стаканом и выпаривают раствор до влажных солей. После охлаждения добавляют от 30 до 40 см3 воды и 16 см3 соляной кислоты, перемешивают до растворения солей, помещают раствор в мерную колбу вместимостью 100 см3 и доводят водой до метки.
Параллельно готовят раствор контрольного опыта с очисткой меди от серебра по 18.3.4.2.
Распыляют полученные растворы в пламени ацетилен-воздух или пропан-бутан-воздух и измеряют поглощение при длине волны 328,1 нм.
Условия измерения подбирают в соответствии с используемым прибором.
Массу серебра в пробе и контрольном опыте устанавливают по градуировочному графику.
18.3.6 Обработка результатов анализа
18.3.6.1 Массовую долю серебра X, %, вычисляют по формуле
|
|
(29) |
где m1 - масса серебра, найденная по градуировочному графику в растворе анализируемой пробы, мг;
т2 - масса серебра, найденная в растворе контрольного опыта, мг;
т - масса навески меди, г.
18.3.6.2 За результат анализа принимают среднеарифметическое значение двух параллельных определений при условии, что абсолютная разность между ними в условиях повторяемости (при доверительной вероятности Р = 0,95) не превышает значений предела повторяемости г, приведенных в таблице 22.
Если расхождение между результатами параллельных определений превышает значение предела повторяемости, выполняют процедуры, изложенные в стандарте [2] (подпункт 5.2.2.1).
18.3.7 Контроль точности результатов анализа
Контроль точности результатов анализа - в соответствии с 3.14.
18.3.8 Оформление результатов анализа
Результаты анализа оформляют в соответствии с 3.15, значения погрешности результатов анализа Д приведены в таблице 22.
18.4 Определение массовой доли марганца, свинца, железа, сурьмы, висмута
18.4.1 Требования к погрешности анализа
Погрешность результатов анализа массовой доли марганца, свинца, железа, сурьмы и висмута, значения пределов повторяемости и воспроизводимости для доверительной вероятности Р = 0,95 должны соответствовать приведенным в таблице 23.
В процентах
|
Определяемый элемент |
Диапазон массовых долей элемента |
Погрешность результатов анализа ±Δ |
Предел |
|
|
повторяемости r (n=2) |
воспроизводимости R |
|||
|
Марганец |
От 0,00020 до 0,00050 включ. |
0,00007 |
0,00005 |
0,00010 |
|
Св. 0,0005 » 0,0010 » |
0,0001 |
0,0001 |
0,0002 |
|
|
» 0,0010 » 0,0020 » |
0,0004 |
0,0003 |
0,0005 |
|
|
» 0,0020 » 0,0050 » |
0,0006 |
0,0006 |
0,0009 |
|
|
Свинец |
От 0,0002 до 0,0005 включ. |
0,0001 |
0,0001 |
0,0002 |
|
Св. 0,0005 » 0,0020 » |
0,0004 |
0,0003 |
0,0005 |
|
|
» 0,0020 » 0,0050 » |
0,0006 |
0,0006 |
0,0009 |
|
|
Железо |
От 0,0002 до 0,0005 включ. |
0,0001 |
0,0001 |
0,0002 |
|
Св. 0,0005 » 0,0010 » |
0,0002 |
0,0002 |
0,0003 |
|
|
» 0,0010 » 0,0020 » |
0,0004 |
0,0003 |
0,0005 |
|
|
» 0,0020 » 0,0050 » |
0,0005 |
0,0005 |
0,0007 |
|
|
Сурьма |
От 0,00030 до 0,00050 включ. |
0,00007 |
0,00005 |
0,00010 |
|
Св. 0,0005 » 0,0010 » |
0,0002 |
0,0002 |
0,0003 |
|
|
» 0,0010 » 0,0030 » |
0,0004 |
0,0004 |
0,0006 |
|
|
» 0,003 » 0,005 » |
0,001 |
0,001 |
0,002 |
|
|
Висмут |
От 0,0002 до 0,0005 включ. |
0,0001 |
0,0001 |
0,0002 |
|
Св. 0,0005 » 0,0010 » |
0,0002 |
0,0002 |
0,0003 |
|
|
» 0,0010 » 0,0020 » |
0,0004 |
0,0003 |
0,0005 |
|
|
» 0,0020 » 0,0050 » |
0,0005 |
0,0006 |
0,0007 |
|
18.4.2 Средства измерений, вспомогательные устройства, материалы, растворы
При выполнении анализа применяют следующие средства измерений, вспомогательные устройства:
- спектрофотометр атомно-абсорбционный с источниками излучения на марганец, свинец, железо, сурьму, висмут;
- компрессор воздушный;
- весы лабораторные специального класса точности по ГОСТ 24104;
- стаканы Н-1-100 ТХС, В-1-250 ТХС, В-1-400 ТХС по ГОСТ 25336;
- колбы конические Кн-2-250-34 ТХС по ГОСТ 25336;
- колбы мерные 2-25-2, 2-50-2, 2-100-2, 2-1000-2 по ГОСТ 1770;
- дефлегматор 250-19/26-29/32 ТС по ГОСТ 25336;
- пипетки 2-2-1-10,2-2-1-5 по ГОСТ 29227;
- пробирки П-2-10-0,1 ХС по ГОСТ 1770.
При выполнении анализа применяют следующие материалы, растворы:
- ацетилен по ГОСТ 5457;
- пропан-бутан по ГОСТ 20448;
- фильтры по [21] или другие средней плотности;
- кислоту азотную особой чистоты по ГОСТ 11125 или кислоту азотную по ГОСТ 4461 (перегнанную в кварцевом аппарате), разбавленную 1:1, 1:4, раствор молярной концентрации 0,1 моль/дм3;
- кислоту соляную особой чистоты по ГОСТ 14261 или кислоту соляную по ГОСТ 3118, разбавленную 1:1, 1:5, 1:10, 7:3;
- кислоту серную по ГОСТ 4204, разбавленную 1:10;
- аммиак водный по ГОСТ 3760, разбавленный 1:19;
- железо по [19], раствор массовой концентрации 15 г/дм3 в азотной кислоте молярной концентрации 0,1 моль/дм3;
- водорода пероксид по ГОСТ 10929, разбавленный 1:10;
- лантан азотнокислый по [22] (хлорид лантана или оксид лантана), раствор массовой концентрации 2 мг/см3;
- висмут по ГОСТ 10928, марка Ви00 или Ви0;
- марганец металлический по ГОСТ 6008;
- свинец высокой чистоты по ГОСТ 22861 или свинец по ГОСТ 3778, марка С00;
- сурьму по ГОСТ 1089, марка Су00;
- воду бидистиллированную или деионизированную;
- бумагу универсальную индикаторную по [14].
18.4.3 Метод анализа
Метод основан на измерении при длинах волн соответственно 279,5; 283,3; 248,3; 217,6; 223,1 нм резонансного излучения атомами марганца, свинца, железа, сурьмы и висмута, образующимися в результате атомизации при введении анализируемого раствора в пламя ацетилен-воздух или пропан-бутан-воздух. Элементы предварительно соосаждают на гидроксиде железа или гидроксиде лантана.
18.4.4 Подготовка к выполнению анализа
18.4.4.1 При приготовлении раствора азотнокислого лантана (оксида лантана или хлорида лантана) массовой концентрации 2 мг/см3 навеску азотнокислого лантана массой 3,1 г (2,4 г оксида лантана или 5,4 г хлорида лантана) растворяют в объеме от 10 до 15 см3 соляной кислоты, разбавленной 1:1. Полученный раствор помещают в мерную колбу вместимостью 1000 см3 и доводят водой до метки.
18.4.4.2 Приготовление растворов известной концентрации
Марганец
Раствор А: навеску марганца массой 0,1000 г растворяют в 20 см3 азотной кислоты, разбавленной 1:1, охлаждают, помещают в мерную колбу вместимостью 1000 см3 и доводят водой до метки.
1 см3 раствора А содержит 0,1 мг марганца.
Раствор Б: 10 см3 раствора А помещают в мерную колбу вместимостью 100 см3, добавляют 1 см3 азотной кислоты, разбавленной 1:1, и доводят водой до метки.
1 см3 раствора Б содержит 0,01 мг марганца.
Раствор В: 20 см3 раствора Б помещают в мерную колбу вместимостью 100 см3, добавляют 3 см3 азотной кислоты, разбавленной 1:1, и доводят водой до метки.
1 см3 раствора В содержит 0,002 мг марганца.
Свинец
Раствор А: навеску свинца массой 0,1000 г растворяют в 10 см3 азотной кислоты, разбавленной 1:1. Раствор помещают в мерную колбу вместимостью 1000 см3 и доводят водой до метки.
1 см3 раствора А содержит 0,1 мг свинца.
Раствор Б: 10 см3 раствора А помещают в мерную колбу вместимостью 100 см3 и доводят водой до метки.
1 см3 раствора Б содержит 0,01 мг свинца.
Раствор В: 20 см3 раствора Б помещают в мерную колбу вместимостью 100 см3 и доводят водой до метки.
1 см3 раствора В содержит 0,002 мг свинца.
Железо
Раствор А: навеску железа массой 0,1000 г растворяют в 20 см3 соляной кислоты, разбавленной 1:1, при нагревании. Раствор охлаждают, помещают в мерную колбу вместимостью 1000 см3 и доводят водой до метки.
1 см3 раствора А содержит 0,1 мг железа.
Раствор Б: 10 см3 раствора А помещают в мерную колбу вместимостью 100 см3, добавляют 10 см3 0,1 М азотной кислоты и доводят водой до метки.
1 см3 раствора Б содержит 0,01 мг железа.
Раствор В: 20 см3 раствора Б помещают в мерную колбу вместимостью 100 см3, добавляют 10 см3 0,1 М азотной кислоты и доводят водой до метки.
1 см3 раствора В содержит 0,002 мг железа.
Сурьма
Раствор А: навеску сурьмы массой 0,1000 г помещают в коническую колбу вместимостью 250 см3, приливают 20 см3 серной кислоты и нагревают до растворения навески. После охлаждения добавляют от 100 до 150 см3 воды, перемешивают, охлаждают, помещают в мерную колбу вместимостью 1000 см3 и доводят до метки серной кислотой, разбавленной 1:10.
При приготовлении раствора А из триоксида сурьмы навеску массой 0,1200 г помещают в коническую колбу вместимостью 1000 см3, снабженную дефлегматором. Приливают 200 см3 соляной кислоты, разбавленной 7:3, и нагревают до растворения навески. После охлаждения раствор упаривают до объема от 5 до 10 см3, помещают в мерную колбу вместимостью 1000 см3 и доводят до метки серной кислотой, разбавленной 1:10.
1 см3 раствора А содержит 0,1 мг сурьмы.
Раствор Б: 10 см3 раствора А помещают в мерную колбу вместимостью 100 см3 и доводят до метки серной кислотой, разбавленной 1:10. Используют свежеприготовленный раствор.
1 см3 раствора Б содержит 0,01 мг сурьмы.
Раствор В: 20 см3 раствора Б помещают в мерную колбу вместимостью 100 см3 и доводят до метки серной кислотой, разбавленной 1:10. Используют свежеприготовленный раствор.
1 см3 раствора В содержит 0,002 мг сурьмы.
Висмут
Раствор А: навеску висмута массой 0,1000 г растворяют при нагревании в 5 см3 соляной кислоты, разбавленной 1:5. После охлаждения приливают от 50 до 80 см3 воды, перемешивают до растворения солей, помещают в мерную колбу вместимостью 1000 см3 и доводят до метки азотной кислотой, разбавленной 1:1.
1 см3 раствора А содержит 0,1 мг висмута.
Раствор Б: 10 см3 раствора А помещают в мерную колбу вместимостью 100 см3 и доводят до метки 0,1 М азотной кислотой.
1 см3 раствора Б содержит 0,01 мг висмута.
18.4.4.3 Построение градуировочных графиков
Для построения градуировочных графиков в ряд стаканов (или конических колб) помещают: 0; 1,0; 2,0; 5,0; 10,0 см3 раствора марганца В и 5,0; 10,0 см3 раствора марганца Б, что соответствует 0; 0,002; 0,004; 0,010; 0,020; 0,050 и 0,100 мг марганца; 0; 1,0; 2,0; 5,0; 10,0 см3 раствора свинца В и 5,0; 10,0 см3 раствора свинца Б, что соответствует 0; 0,002; 0,004; 0,010; 0,020; 0,050; 0,100 мг свинца; 0; 1,0; 2,0; 5,0; 10,0 см3 раствора железа В и 5,0; 10,0 см3 раствора железа Б, что соответствует 0; 0,002; 0,004; 0,010; 0,020; 0,050; 0,10 мг железа; 0; 1,0; 3,0; 5,0 см3 раствора сурьмы В и 3,0; 5,0; 10,0 см3 раствора сурьмы Б, что соответствует 0; 0,002; 0,006; 0,010; 0,030; 0,050; 0,100 мг сурьмы; 0; 0,5; 1,0; 2,5; 5,0; 7,5; 10,0 см3 раствора висмута Б, что соответствует 0; 0,005; 0,010; 0,025; 0,050; 0,075; 0,100 мг висмута.
Во все стаканы (или колбы) приливают по 3 см3 раствора железа или лантана (в стаканы, содержащие растворы железа известной концентрации, приливают по 3 см3 раствора лантана), добавляют от 5 до 10 см3 азотной кислоты, разбавленной 1:1, от 3 до 5 см3 раствора пероксида водорода, от 80 до 100 см3 воды и нагревают раствор до кипения. В раствор добавляют аммиак до выпадения осадка гидроксида железа или лантана и еще дополнительно 5 см3. Выдерживают раствор с осадком при температуре от 60°С до 70°С до коагуляции осадка. Затем фильтруют через фильтр средней плотности «белая лента» и промывают осадок на фильтре 4 - 5 раз горячим водным раствором аммиака, разбавленным 1:19.
Осадок на фильтре растворяют в объеме от 10 до 15 см3 горячей соляной кислоты, разбавленной 1:1 (осадок гидроксида со свинцом растворяют в горячей азотной кислоте, разбавленной 1:1). Промывают фильтр горячей водой до нейтральной реакции промывных вод (по универсальной индикаторной бумаге), собирая фильтрат в стакан (или колбу), в котором проводили осаждение. Упаривают раствор до объема от 6 до 8 см3, охлаждают, помещают раствор в мерную пробирку вместимостью 10 см3 или мерную колбу вместимостью 25 см3 в зависимости от массы элементов и доводят до метки водой.
Распыляют растворы в пламени ацетилен-воздух или пропан-бутан-воздух и измеряют поглощение резонансных линий элементов при длинах волн: марганца - 279,5 нм, свинца - 283,3 нм, железа - 248,3 нм, сурьмы - 217,6 нм, висмута - 223,1 нм.
По полученным значениям оптических плотностей и соответствующим массовым концентрациям элементов в растворах известной концентрации строят градуировочные графики в прямоугольных координатах, используя для каждой точки среднее значение из двух измерений оптической плотности.
18.4.5 Выполнение анализа
18.4.5.1 Для удаления частиц железа (возможно занесенных при подготовке пробы к анализу) стружку меди обрабатывают магнитом. Затем стружку обмывают от 5 до 10 см3 соляной кислоты, разбавленной 1:10, и дважды бидистиллированной (или деионизированной) водой.
18.4.5.2 Навеску меди массой 2,0000 г помещают в стакан (или коническую колбу) вместимостью 250 см3 (параллельно ведется контрольный опыт) и приливают от 20 до 25 см3 азотной кислоты, разбавленной 1:1. Нагревают до растворения навески. Затем приливают 100 см3 воды, 10 см3 раствора лантана, от 3 до 5 см3 раствора пероксида водорода и нагревают раствор до кипения. В раствор добавляют раствор аммиака до появления не исчезающей при перемешивании интенсивно синей окраски раствора аммиачного комплекса меди и еще дополнительно 5 см3. Выдерживают стакан (или колбу) в теплом месте плиты до коагуляции осадка.
Фильтруют раствор через фильтр средней плотности и промывают осадок на фильтре 4-5 раз горячим раствором аммиака, разбавленным 1:19. Затем осадок на фильтре растворяют в объеме от 10 до 15 см3 горячей азотной кислоты, разбавленной 1:1, и промывают фильтр горячей водой до нейтральной реакции промывных вод (по универсальной индикаторной бумаге), собирая фильтрат в стакан (колбу), в котором проводили осаждение. Раствор упаривают до объема от 6 до 8 см3, охлаждают, помещают в мерную пробирку вместимостью 10 см3 или в мерную колбу вместимостью 25 см3 в зависимости от массовой доли элементов и доводят водой до метки.
Распыляют анализируемые растворы и растворы контрольного опыта в пламени ацетилен-воздух или пропан-бутан-воздух и измеряют поглощение резонансных линий элементов. Условия измерения подбирают в соответствии с используемым прибором. Массовую концентрацию элементов определяют по градуировочным графикам.
18.4.6 Обработка результатов анализа
18.4.6.1 Массовую долю элемента X, %, вычисляют по формуле
|
|
(30) |
где m1 - концентрация определяемого элемента в растворе пробы, найденная по градуировочному графику, мг/см3;
т2 - концентрация определяемого элемента в растворе контрольного опыта, мг/см3;
V - вместимость мерной колбы (пробирки), см3;
т - масса навески меди, г.
18.4.6.2 За результат анализа принимают среднеарифметическое значение двух параллельных определений при условии, что абсолютная разность между ними в условиях повторяемости (при доверительной вероятности Р - 0,95) не превышает значений предела повторяемости г, приведенных в таблице 23.
Если расхождение между результатами параллельных определений превышает значение предела повторяемости, выполняют процедуры, изложенные в стандарте [2] (подпункт 5.2.2.1).
18.4.7 Контроль точности результатов анализа
Контроль точности результатов анализа проводят в соответствии с 3.14.
18.4.8 Оформление результатов анализа
Результаты анализа оформляют в соответствии с 3.15, значения погрешности результатов анализа А приведены в таблице 23.
18.5 Определение массовой доли селена и теллура
18.5.1 Определение массовой доли селена
18.5.1.1 Требования к погрешности анализа
Погрешность результатов анализа массовой доли селена, значения пределов повторяемости и воспроизводимости для доверительной вероятности Р = 0,95 должны соответствовать приведенным в таблице 24.
В процентах
|
Диапазон массовой доли селена |
Погрешность результатов анализа ±Δ |
Предел |
|
|
повторяемости r (n=2) |
воспроизводимости R |
||
|
От 0,00002 до 0,00005 включ. |
0,00002 |
0,00002 |
0,00003 |
|
Св. 0,00005 » 0,00010 » |
0,00004 |
0,00004 |
0,00005 |
|
» 0,00010 » 0,00030 » |
0,00006 |
0,00006 |
0,00009 |
|
» 0,00030 » 0,00050 » |
0,00011 |
0,00010 |
0,00014 |
18.5.1.2 Средства измерений, вспомогательные устройства, материалы, растворы
При выполнении анализа применяют следующие средства измерений, вспомогательные устройства:
- спектрофотометр атомно-абсорбционный, модель 503 фирмы Перкин-Эльмер или другой фирмы с источником излучения на селен;
- атомизатор электротермический марки HGA-76 или другой аналогичный;
- весы лабораторные специального класса точности по ГОСТ 24104;
- стаканы Н-1-100 ТХС по ГОСТ 25336;
- колбы мерные 2-50-2, 2-100-2 по ГОСТ 1770;
- пипетки 5-2-1-1, 5-2-1-2, 5-2-1-5 по ГОСТ 29227;
- цилиндр 1-10 по ГОСТ 1770;
- пробирки П-2-10-0,2 ХС, П-2-20-0,2 ХС, П-2-25-0,2 ХС по ГОСТ 1770;
- микродозатор (микропипетка) вместимостью от 0,01 до 0,02 см3.
При выполнении анализа применяют следующие материалы, растворы:
- аргон газообразный высшего сорта по ГОСТ 10157;
- воздух сжатый под давлением от 2·105 до 6·105 Па (от 2 до 6 кгс/см2);
- селен по ГОСТ 10298;
- кислоту азотную по ГОСТ 4461, разбавленную 1:1;
- кислоту соляную по ГОСТ 3118;
- кислоту бромисто-водородную по ГОСТ 2062;
- кислоту хлорную по [23], раствор с массовой долей 57%;
- толуол по ГОСТ 5789;
- спирт этиловый по ГОСТ 18300.
18.5.1.3 Метод анализа
Определение селена проводят атомно-абсорбционным методом с электротермическим атомизатором (ЭТА) по аналитической линии 196,0 нм после растворения навески меди в азотной кислоте и экстракционного концентрирования селена толуолом из анализируемого раствора, содержащего 8 моль/дм3 соляной кислоты, 2 моль/дм3хлорной кислоты, 0,2 моль/дм3 бромисто-водородной кислоты и 50 г/см3 меди.
18.5.1.4 Подготовка к проведению анализа
Приготовление растворов сравнения (PC)
Раствор А: навеску металлического селена массой 0,1000 г растворяют в смеси от 10 до 15 см3 соляной кислоты и от 0,1 до 0,2 см3 азотной кислоты, нагревая на водяной бане до полного растворения селена. Охлажденный раствор помещают в мерную колбу вместимостью 100 см3, доливают до метки соляной кислотой, разбавленной 1:1.
1 см3 раствора А содержит 1 мг селена.
Раствор Б: 2 см3 раствора А помещают в стакан вместимостью 100 см3, приливают 10 см3 хлорной кислоты и нагревают на водяной бане до появления паров хлорной кислоты. Стакан снимают с водяной бани, охлаждают, обмывают стенки стакана водой и повторяют выпаривание до паров хлорной кислоты. Раствор помещают в мерную колбу вместимостью 100 см3, доводят до метки соляной кислотой.
1 см3 раствора Б содержит 20 мкг селена.
Раствор В: 1 см3 раствора Б помещают в пробирку вместимостью 25 см3, приливают 2 см3 хлорной кислоты, доводят до метки соляной кислотой, добавляют 0,3 см3 бромисто-водородной кислоты, 10 см3 толуола. Экстрагируют 15 мин.
1 см3 раствора В содержит 2 мкг селена.
Приготовление растворов сравнения для атомно-абсорбционного анализа (не менее трех)
Раствор Г: 1 см3 раствора В помещают в пробирку вместимостью 10 см3 и добавляют 9 см3 толуола.
1 см3 раствора Г содержит 0,2 мкг селена.
В две пробирки вместимостью 10 см3 каждая помещают по 1 см3 раствора Г, приливают в одну 3 см3, в другую 1 см3 толуола. Растворы имеют, соответственно, концентрацию селена 0,05 и 0,1 мкг/см3.
Приготовление синтетической смеси для проверки правильности методики и правильности работы прибора
Отбирают 2,5 см3 раствора Б, приготовленного по 18.5.1.4, помещают в мерную колбу вместимостью 100 см3 и доводят водой до метки. Погрешность введенного содержания селена - не более 2%. Смесь готовят перед использованием.
1 см3 смеси содержит (0,5 ±0,02) мкг селена.
Подготовка измерительного прибора
Включение и настройку прибора и ЭТА проводят в соответствии с инструкцией по эксплуатации.
Правильность работы прибора и положение градуировочного графика проверяют, подвергая анализу синтетическую смесь, приготовленную в соответствии с 18.5.1.4. Аналитический сигнал при введении 0,02 см3 раствора, содержащего 0,5 мкг/см3 селена, должен быть около 0,2 единиц абсорбции.
Используют аналитическую линию 196,0 нм, щель 2,0 нм и режим работы ЭТА, приведенный в таблице 25.
|
Стадии термической обработки проб |
Температура, °С |
Время, с |
|
Сушка |
150 |
10 |
|
Разложение |
700 - 900 |
10 |
|
Атомизация |
2400 |
5 |
|
Примечание - При работе с аналогичным прибором другой марки условия проведения анализа корректируют, исходя из обеспечения максимального аналитического сигнала селена при анализе одного из растворов сравнения. |
||
При использовании приборов других марок аналитический сигнал при введении синтетической смеси должен соответствовать значению, приведенному в паспорте на данный прибор.
Микропипеткой вместимостью 0,02 см3 вводят в ЭТА толуол и варьированием температуры разложения от 700°С до 900°С добиваются того, чтобы абсорбционный сигнал толуола был не больше, чем при простом обжиге кюветы. Эту операцию выполняют для каждой графитовой трубки.
18.5.1.5 Выполнение анализа
Навеску образца меди массой 1,0000 г помещают в стакан вместимостью 100 см3, приливают от 10 до 15 см3 азотной кислоты, разбавленной 1:1, и нагревают до растворения навески. Упаривают раствор до объема от 5 до 6 см3, приливают 7 см3 хлорной кислоты. Нагревают до появления паров хлорной кислоты и убирают стакан с плиты; охлаждают, добавляют 3 см3 воды и снова нагревают до появления белых паров. Раствор охлаждают до температуры от 30°С до 50°С, приливают 10 см3 соляной кислоты, нагревают до температуры не более 50°С, перемешивая до полного или частичного растворения солей. Содержимое переводят в пробирку вместимостью 20 см3 (или 25 см3), обмывают стакан от 0,5 до 1 см3 воды и соляной кислотой, добавляют в пробирку 0,6 см3 бромисто-водородной кислоты и доводят до метки соляной кислотой. Перемешивают до растворения осадка, открывая пробирку каждый раз после 2 - 4 встряхиваний.
В пробирку вместимостью 10 см3 помещают 2 см3 толуола и 2 см3 раствора пробы (если ожидаемая массовая доля селена от 2·10-4 % до 5·10-4 %, соотношение объемов органической и водной фаз 1:1) или 4 см3 раствора пробы (если ожидаемая массовая доля селена менее 2·10-4 %, соотношение фаз 1:2) и экстрагируют 15 мин. Экстракт должен быть бесцветным, водная фаза - темно-коричневой. Фазы не делят; для анализа используют экстракт.
Аликвотные части (0,02 см3) растворов сравнения, анализируемой пробы и контрольного опыта последовательно вводят в ЭТА. Измеряют величину сигнала абсорбции селена. Для каждого раствора делают 2 - 3 измерения. Через каждые 5 - 8 проб анализируют один из растворов сравнения. Периодически, по истечении от 2 до 3 ч работы, стекла атомизатора и графитовые контакты ЭТА протирают ватой, смоченной спиртом. После чистки проводят обжиг графитовой трубки.
18.5.1.6 Обработка результатов анализа
Массовую долю селена X, %, вычисляют по формуле
|
X = KhF, |
(31) |
где К - коэффициент пересчета, равный: 1·10-3 (при соотношении объемов фаз 1:2) или 2·10-3 (при соотношении объемов фаз 1:1);
h - значение абсорбции селена в растворе анализируемой пробы, мм;
F - градуировочный фактор, равный
|
|
(32) |
где п - количество растворов сравнения;
Сi - концентрация селена в i-м растворе сравнения, мкг/см3;
Hi -значение абсорбции селена в i-м растворе сравнения, мм.
Массовую долю селена в анализируемой пробе Х в процентах можно также определить по градуировочному графику. На оси абсцисс откладывают массовые доли селена в растворах сравнения, умноженные на 1·10-3 (при соотношении фаз 1:2) или на 2·10-3 (при соотношении фаз 1:1), на оси ординат - среднеарифметические значения высот соответствующих им пиков абсорбции.
За результат анализа принимают среднеарифметическое значение двух параллельных определений при условии, что абсолютная разность между ними в условиях повторяемости не превышает значений (при доверительной вероятности Р = 0,95) предела повторяемости г, приведенных в таблице 24.
Если расхождение между результатами параллельных определений превышает значение предела повторяемости, выполняют процедуры, изложенные в стандарте [2] (подпункт 5.2.2.1).
18.5.1.7 Контроль точности результатов анализа
Контроль точности результатов анализа - в соответствии с 3.14.
18.5.1.8 Оформление результатов анализа
Результаты анализа оформляют в соответствии с 3.15, значения погрешности результатов анализа Δ приведены в таблице 24.
18.5.2 Определение теллура
18.5.2.1 Требования к погрешности анализа
Погрешность результатов анализа массовой доли теллура, значения пределов повторяемости и воспроизводимости для доверительной вероятности Р = 0,95 должны соответствовать приведенным в таблице 26.
В процентах
|
Диапазон массовой доли теллура |
Погрешность результатов анализа ±Δ |
Предел |
|
|
повторяемости r (n=2) |
воспроизводимости R |
||
|
От 0,00001 до 0,00002 включ. |
0,00001 |
0,00001 |
0,00002 |
|
Св. 0,00002 » 0,00005 » |
0,00002 |
0,00002 |
0,00003 |
|
» 0,00005 » 0,00020 » |
0,00004 |
0,00004 |
0,00005 |
18.5.2.2 Средства измерений, вспомогательные устройства, материалы, растворы
При выполнении анализа применяют следующие средства измерений, вспомогательные устройства:
- спектрофотометр атомно-абсорбционный, модель 503 фирмы Перкин-Эльмер или другой фирмы с источником излучения на теллур;
- атомизатор электротермический марки HGA-76 или другой аналогичный;
- весы лабораторные специального класса точности по ГОСТ 24104;
- стаканы Н-1-100 ТХС по ГОСТ 25336;
- колбы мерные 2-50-2, 2-100-2 по ГОСТ 1770;
- пипетки 5-2-1-2, 5-2-1-5 по ГОСТ 29227;
- пробирки П-2-10-0,2 ХС по ГОСТ 1770;
- микродозатор (микропипетка) вместимостью от 0,01 до 0,02 см3;
- цилиндры 1-10,1-50 по ГОСТ 1770;
- стекло часовое.
При выполнении анализа применяют следующие материалы, растворы:
- аргон газообразный высшего сорта по ГОСТ 10157;
- воздух, сжатый под давлением от 2·105 до 6·105 Па (от 2 до 6 кгс/см2);
- теллур высокой чистоты марки Т-А1 по [27];
- кислоту азотную по ГОСТ 4461, разбавленную 1:1;
- кислоту соляную по ГОСТ 3118;
- толуол по ГОСТ 5789;
- спирт этиловый по ГОСТ 18300;
- водорода пероксид по ГОСТ 10929, стабилизированный продукт;
- триалкилбензиламмоний хлорид (ТАБАХ), экстрагент - раствор молярной концентрации 0,05 моль/дм3 ТАБАХ в толуоле.
18.5.2.3 Метод анализа
Определение теллура проводят атомно-абсорбционным методом с электротермическим атомизатором (ЭТА) по длине волны 214,3 нм после растворения навески меди в соляной кислоте с пероксидом водорода и экстракционного концентрирования теллура раствором 0,05 моль/дм3 триалкилбензиламмоний хлорида в толуоле из анализируемого раствора, содержащего от 3 до 6 моль/дм3 соляной кислоты и 50 г/см3 меди.
18.5.2.4 Подготовка к проведению анализа
Приготовление экстрагента
При приготовлении раствора триалкилбензиламмония хлорида молярной концентрации 0,05 моль/дм3 в толуоле в сухую емкость объемом не менее 1 дм3 переносят 42 см3 технического ТАБАХ и 960 см3 толуола, обмывая им мерный стакан (цилиндр) из-под ТАБАХ.
Приготовление растворов сравнения
Раствор А: навеску металлического теллура массой 0,0100 г помещают в стакан и растворяют в объеме от 5 до 10 см3 азотной кислоты, разбавленной 1:1, нагревая до полного разложения навески. В мерную колбу вместимостью 100 см3 наливают 50 см3 соляной кислоты, переливают охлажденный раствор из стакана, обмывая его водой, и доводят водой до метки.
1 см3 раствора А содержит 0,1 мг теллура.
Раствор Б: 1,0 см3 раствора А помещают в мерную колбу вместимостью 50 см3 и доводят до метки соляной кислотой, разбавленной 1:1.
1 см3 раствора Б содержит 2 мкг теллура.
Раствор В: 2,5 см3 раствора Б помещают в мерную колбу вместимостью 100 см3 и доводят до метки соляной кислотой, разбавленной 1:1.
1 см3 раствора В содержит 0,05 мкг теллура.
Приготовление растворов сравнения для атомно-абсорбционного анализа (не менее трех): в три мерные пробирки вместимостью 10 см3 каждая помещают 1, 2 и 4 см3 раствора В, приливают по 0,4 см3 экстракта любой из проб (приготовленной по 18.4.2.5), доводят соляной кислотой, разбавленной 1:1, до объема 4 см3 и добавляют 2 см3 экстрагента. Экстрагируют 15 мин.
Экстракты содержат 0,025; 0,05 и 0,1 мкг/см3 теллура.
Приготовление синтетической смеси для проверки правильности методики и правильности работы прибора: 5 см3 раствора Б помещают в мерную колбу вместимостью 50 см3 и доводят до метки водой. Погрешность введения теллура - не более 2%.
1 см3 смеси содержит (0,200 ±0,004) мкг теллура.
Подготовка измерительного прибора
Включение и настройку прибора и ЭТА проводят в соответствии с инструкцией по эксплуатации.
Проверяют правильность работы прибора, используя смесь, приготовленную, как описано выше.
Используют аналитическую линию 214,3 нм, щель 0,2 нм и режим работы ЭТА, приведенный в таблице 27.
Микропипеткой вместимостью 0,02 см3 вводят в ЭТА раствор экстрагента в толуоле и варьированием температуры разложения от 800°С до 1000°С добиваются того, чтобы абсорбционный сигнал раствора был не больше, чем при обжиге кюветы. Эту операцию выполняют для каждой графитовой трубки.
|
Стадии термической обработки проб |
Температура, °С |
Время, с |
|
Сушка |
150 |
10 |
|
Разложение |
800 - 1000 |
8 |
|
Атомизация |
2500 |
5 - 7 |
|
Примечание - При работе с аналогичным прибором другой марки условия проведения анализа корректируют, исходя из обеспечения максимального аналитического сигнала теллура при анализе одного из растворов сравнения. |
||
18.5.2.5 Выполнение анализа
Навеску образца меди массой 0,5000 г помещают в стакан вместимостью 100 см3, пипеткой добавляют 4,2 см3 соляной кислоты, накрывают часовым стеклом и добавляют 3 см3 раствора пероксида водорода. Для ускорения реакции смесь взбалтывают 3 - 5 раз. После прекращения реакции (выделение пузырьков) по истечении от 5 до 8 мин добавляют еще 4 см3 пероксида водорода, взбалтывают 3 - 5 раз. После растворения навески меди стакан ставят на плиту, раствор доводят до кипения и по истечении от 2 до 3 мин после разложения избытка пероксида водорода стакан снимают с плиты, охлаждают и доводят объем до 100 см3 водой.
В пробирку вместимостью 10 см3 вносят 2 см3 экстрагента и 2 см3 раствора пробы для ожидаемых массовых долей теллура от 1·10-4 % до 2·10-4 % (соотношение объемов органической и водной фаз при этом - 1:1) или 4 см3 раствора пробы для ожидаемых массовых долей теллура менее 1·10-4 % (соотношение фаз - 1:2). Экстрагируют 15 мин. Фазы не делят, для анализа используют экстракт.
Аликвотные части растворов сравнения, проб и раствора контрольного опыта объемом 0,02 см3 последовательно вводят в ЭТА. Измеряют величину сигнала абсорбции теллура. Для каждого раствора делают 2 - 3 измерения сигнала. Через каждые 5 - 8 проб анализируют один из растворов сравнения. Стекла ЭТА по истечении от 2 до 3 ч протирают ватой, смоченной спиртом, а затем насухо хлопчатобумажной тканью. В конце работы графитовые контакты ЭТА протирают ватой, смоченной спиртом.
18.5.2.6 Обработка результатов анализа
Массовую долю теллура X, %, вычисляют по формуле
|
X = KhF, |
(33) |
где К - коэффициент пересчета, равный: 1·10-3 (при соотношении объемов фаз 1:2) или 2·10-3 (при соотношении объемов фаз 1:1);
h - значение абсорбции теллура в растворе анализируемой пробы, мм;
F - градуировочный фактор, равный
|
|
(34) |
где п - количество растворов сравнения;
Ci - концентрация теллура в i-м растворе сравнения, мкг/см3;
Hi -значение абсорбции теллура в i-м растворе сравнения, мм.
Массовую долю теллура в пробе X в процентах можно также найти по градуировочному графику, на оси абсцисс которого откладывают массовые доли теллура в растворах сравнения, умноженные на 2·10-3 (при соотношении фаз 1:1) или на 1·10-3 (при соотношении фаз 1:2), а на оси ординат - среднеарифметические значения высот соответствующих им пиков абсорбции.
За результат анализа принимают среднеарифметическое значение двух параллельных определений при условии, что абсолютная разность между ними в условиях повторяемости не превышает значений (при доверительной вероятности Р = 0,95) предела повторяемости r, приведенных в таблице 26.
Если расхождение между результатами параллельных определений превышает значение предела повторяемости, выполняют процедуры, изложенные в стандарте [2] (подпункт 5.2.2.1).
18.5.2.7 Контроль точности результатов анализа
Контроль точности результатов анализа проводят в соответствии с 3.14.
18.5.2.8 Оформление результатов анализа
Результаты анализа оформляют в соответствии с 3.15, значения погрешности результатов анализа А приведены в таблице 26.
18.6 Определение массовой доли висмута, олова и серебра
18.6.1 Требования к погрешности анализа
Погрешность результатов анализа массовой доли висмута, олова и серебра, значения пределов повторяемости и воспроизводимости для доверительной вероятности Р = 0,95 должны соответствовать приведенным в таблице 28.
В процентах
|
Наименование элемента |
Диапазон массовых долей элемента |
Погрешность результатов анализа ±∆ |
Предел |
|
|
повторяемости r (n=2) |
воспроизводимости R |
|||
|
Висмут |
От 0,000010 до 0,000020 включ. |
0,000007 |
0,000008 |
0,000012 |
|
Св. 0,00002 » 0,00005 » |
0,00001 |
0,00001 |
0,00002 |
|
|
» 0,00005 » 0,00010 » |
0,00002 |
0,00002 |
0,00003 |
|
|
» 0,00010 » 0,00050 » |
0,00004 |
0,00005 |
0,00006 |
|
|
Олово |
От 0.00001 до 0,00002 включ. |
0,00001 |
0,00001 |
0,00002 |
|
» 0,00002 » 0,00005 » |
0,00002 |
0,00001 |
0,00003 |
|
|
» 0,00005 » 0,00010 » |
0,00002 |
0,00002 |
0,00004 |
|
|
» 0,00010 » 0,00050 » |
0,00004 |
0,00007 |
0,00007 |
|
|
Серебро |
От 0,00020 до 0,00050 включ. |
0,00008 |
0,00010 |
0,00013 |
|
» 0,00050 » 0,00100 » |
0,00012 |
0,00014 |
0,00020 |
|
|
» 0,0010 » 0,0030 » |
0,0002 |
0,0002 |
0,0004 |
|
18.6.2 Средства измерений, вспомогательные устройства, материалы, растворы
При выполнении анализа применяют следующие средства измерений, вспомогательные устройства:
- спектрофотометр атомно-абсорбционный фирмы «Хитачи» с атомизатором электротермическим или фирмы Перкин-Эльмер, модель 403, или другой фирмы с источниками излучения на висмут, олово, серебро;
- аппарат для перемешивания жидкости типа АВБ-4П;
- весы лабораторные специального класса точности по ГОСТ 24104;
- пробирки П-2-100-0,2 ХС по ГОСТ 1770;
- воронки делительные ВД-1-1000 по ГОСТ 25336;
- колбы мерные 2-50-2, 2-100-2, 1-500-2, 2-1000-2 по ГОСТ 1770;
- пипетки 1-2-1-1, 1-2-1-2, 2-2-2-5, 2-2-2-10, 2-2-2-25, 2-2-2-50, 4-2-1-1, 4-2-1-2, 5-2-2-2 по ГОСТ 29227;
- стаканы В-1-100 ТС по ГОСТ 25336;
- микродозатор (микропипетка) вместимостью от 0,01 до 0,02 см3.
При выполнении анализа применяют следующие материалы, растворы:
- бумагу индикаторную универсальную по [14];
- кислоту азотную особой чистоты по ГОСТ 11125, разбавленную 1:1; -кислоту серную особой чистоты по ГОСТ 14262, разбавленную 1:1 и 1:17;
- кислоту соляную особой чистоты по ГОСТ 14261, разбавленную 1:5, 1:10, 1:100;
- аммиак водный по ГОСТ 3760, разбавленный 1:1;
- серебро по ГОСТ 6836;
- олово марки 01 по ГОСТ 860;
- висмут марки Ви00 по ГОСТ 10928;
- спирт этиловый по ГОСТ 18300;
- толуол по ГОСТ 5789;
- ацетилен по ГОСТ 5457;
- аргон по ГОСТ 10157;
- катионит КУ-2-8 по ГОСТ 20298;
- триалкилбензоламмоний хлорид (ТАБАХ), 1,3 моль/дм3; раствор молярной концентрации 0,26 моль/дм3 в толуоле;
- воду дистиллированную по ГОСТ 6709, деионизированную, полученную пропусканием через ионообменную колонку с катионитом, например КУ-2-8;
- средства моющие синтетические порошкообразные по ГОСТ 25644.
18.6.3 Метод анализа
Метод включает кислотное разложение навески пробы, экстракционное выделение висмута, олова, серебра раствором ТАБАХ в толуоле и последующее атомно-абсорбционное определение массовой доли висмута, олова, серебра в органической фазе. Атомизация проб при определении массовой доли висмута и олова осуществляется в электротермическом атомизаторе (ЭТА), при определении массовой доли серебра - в пламени ацетилен-воздух.
18.6.4 Подготовка к выполнению анализа
18.6.4.1 Приготовление раствора ТАБАХ молярной концентрации 0,26 моль/дм3 в толуоле
ТАБАХ (1,3 моль/дм3) разбавляют толуолом в соотношении 1:4, помещают в делительную воронку и дважды промывают в течение от 4 до 5 мин равным объемом аммиака, разбавленного 1:1. Отстаивают до полного разделения фракций. Затем промывают водой, соляной кислотой, разбавленной 1:5, и дважды соляной кислотой, разбавленной 1:100. Проверяют кислотность водной фазы, рН раствора должен быть равен от 1 до 2. При рН больше 2 повторяют промывку соляной кислотой, разбавленной 1:5, затем соляной кислотой, разбавленной 1:100.
18.6.4.2 Приготовление растворов известной концентрации
Приготовление раствора висмута массовой концентрации 0,1 мг/см3/
Навеску металлического висмута массой 0,1000 г помещают в стакан вместимостью 100 см3, растворяют в 10 см3 азотной кислоты, разбавленной 1:1, выпаривают с 5 см3 серной кислоты до белых паров, охлаждают, помещают в мерную колбу вместимостью 1000 см3 и доводят до метки серной кислотой, разбавленной 1:17.
Приготовление раствора олова массовой концентрации 0,1 мг/см3
Навеску металлического олова массой 0,1000 г помещают в стакан вместимостью 100 см3, растворяют в 20 см3 соляной кислоты при нагревании на песчаной бане, не доводя до кипения, охлаждают, помещают в мерную колбу вместимостью 1000 см3 и доводят до метки водой.
Приготовление раствора серебра массовой концентрации 0,1 мг/см3
Навеску металлического серебра массой 0,1000 г помещают в стакан вместимостью 100 см3, растворяют в 10 см3 азотной кислоты, разбавленной 1:1, помещают в мерную колбу вместимостью 1000 см3 и доводят до метки водой.
18.6.4.3 Приготовление рабочих растворов известной концентрации
Приготовление рабочих растворов висмута и олова
В мерную колбу вместимостью 500 см3 вносят по 5 см3 растворов висмута и олова и доводят до метки соляной кислотой, разбавленной 1:10. Затем пипеткой отбирают 50 см3 раствора, помещают в пробирку вместимостью 100 см3 (или в делительную воронку) и проводят экстракцию равным объемом раствора ТАБАХ в течение 30 мин на аппарате для перемешивания жидкости.
1 см3 экстракта содержит по 1 мкг висмута и олова.
В мерные колбы вместимостью 25 см3 каждая вносят по 0,50; 1,25; 2,50; 5,00; 12,50 см3 экстракта и доводят до метки раствором ТАБАХ. Рабочие растворы содержат по 0,02; 0,05; 0,10; 0,20; 0,50 мкг/см3 висмута и олова.
Приготовление рабочих растворов серебра
В мерную колбу вместимостью 500 см3 вносят 50 см3 раствора серебра известной концентрации и доводят до метки соляной кислотой, разбавленной 1:10. Пипеткой отбирают 50 см3 раствора, помещают в пробирку вместимостью 100 см3 (или в делительную воронку) и проводят экстракцию равным объемом раствора ТАБАХ в течение 30 мин на аппарате для перемешивания жидкости. Полученный экстракт содержит 10 мкг/см3 серебра.
В мерные колбы вместимостью 50 см3 каждая вносят 0,50; 1,25; 2,50; 5,00; 10,00; 15,00 см3 экстракта и доводят до метки раствором ТАБАХ. Рабочие растворы содержат 0,10; 0,25; 0,50; 1,00; 2,00; 3,00 мкг/см3 серебра.
Рабочие растворы устойчивы в течение четырех дней.
18.6.4.4 Построение градуировочных графиков
Для построения градуировочных графиков измеряют абсорбцию рабочих растворов в начале и в конце фотометрирования партии проб, и по усредненным значениям величин абсорбции и соответствующим им массовым концентрациям строят градуировочные графики в прямоугольных координатах.
Фотометрирование каждого раствора проводят не менее двух раз.
18.6.4.5 Подготовка приборов к анализу
Условия проведения анализа и подготовительные работы, необходимые для приведения спектрофотометров в рабочее состояние, - в соответствии с инструкциями по эксплуатации прибора.
Стадии и условия процесса атомизации пробы в графитовой кювете приведены в таблице 29.
Таблица 29 - Температурный режим для электротермического атомизатора
|
Элемент |
Стадия |
Начальная температура, °С |
Конечная температура, °С |
Время, с |
|
Висмут |
Сушка |
50 |
120 |
30 |
|
Озоление |
120 |
400 |
10 |
|
|
Озоление |
400 |
550 |
20 |
|
|
Атомизация |
1800 |
1800 |
7 |
|
|
Очистка, продувка кюветы |
2400 |
2400 |
10 |
|
|
Олово |
Сушка |
25 |
100 |
10 |
|
Сушка |
100 |
120 |
10 |
|
|
Озоление |
120 |
400 |
10 |
|
|
Озоление |
400 |
400 |
10 |
|
|
Атомизация |
2700 |
2700 |
7 |
|
|
Очистка, продувка кюветы |
2800 |
2800 |
3 |
18,6.5 Выполнение анализа
18.6.5.1 Навеску пробы массой 1,0000 г помещают в стакан вместимостью 100 см3 и растворяют при нагревании в 15 см3 азотной кислоты, разбавленной 1:1, упаривают до влажных солей. Приливают 7 см3 серной кислоты, разбавленной 1:1, и упаривают до появления паров серной кислоты. Остаток охлаждают и растворяют при нагревании в 10 см3 соляной кислоты, разбавленной 1:100, переносят в мерную колбу вместимостью 50 см3, обмывают стакан от 5 до 7 см3 соляной кислоты, разбавленной 1:100, и присоединяют к раствору в мерной колбе. Пипеткой добавляют 2 см3 раствора ТАБАХ и проводят экстракцию в течение 30 мин на аппарате для перемешивания. После полного расслоения органическую фазу поднимают в узкую часть колбы добавлением 20 см3 соляной кислоты, разбавленной 1:100.
18.8.5.2 При определении массовой концентрации серебра капилляр опускают в колбу с экстрактом и проводят фотометрирование по 18.6.4.5. Капилляр и распылительную систему после каждого фотометрирования промывают раствором ТАБАХ в течение 10 - 15 с, В конце фотометрирования партии проб капилляр и распылительную систему промывают объемом от 8 до 10 см3 этилового спирта.
18.6.5.3 Микропипеткой переносят 0,02 см3 анализируемого раствора висмута или олова в дважды обожженную графитовую кювету и фотометрируют по 18.6.4.5.
После 8 - 10 измерений осуществляют операцию обжига и продувки кюветы.
18.6.5.4 Одновременно с проведением анализа проводят контрольный опыт для внесения в результат анализа поправки, учитывающей массовую долю определяемых элементов в реактивах и материалах. Поправку вычисляют как среднеарифметическое значение трех параллельных определений.
18.6.6 Обработка результатов анализа
18.6.6.1 Массовую долю висмута, олова и серебра X, %, вычисляют по формуле
|
|
(35) |
где С - массовая концентрация элемента, найденная по градуировочному графику, мкг/см3;
СK - массовая концентрация элемента в растворе контрольного опыта, мкг/см3;
V - объем фотометрируемого экстракта, см3;
т - масса навески пробы, г.
18.6.6.2 За результат анализа принимают среднеарифметическое значение двух параллельных определений при условии, что абсолютная величина разности между ними в условиях повторяемости не превышает значений (при доверительной вероятности Р = 0,95) предела повторяемости г, приведенных в таблице 28.
Если расхождение между результатами параллельных определений превышает значение предела повторяемости, выполняют процедуры, изложенные в стандарте [2] (подпункт 5.2.2.1).
18.6.7 Контроль точности результатов анализа
Контроль точности результатов анализа - в соответствии с 3.14.
18.6.8 Оформление результатов анализа
Результаты анализа оформляют в соответствии с 3.15, значения погрешности результатов анализа Δ приведены в таблице 28.
19 Метод спектрального анализа по оксидным стандартным образцам с фотоэлектрической регистрацией спектра
19.1 Область применения
В настоящем разделе установлено определение массовой доли примесей в меди в диапазонах, приведенных в таблице 30, методом спектрального анализа по оксидным образцам с фотоэлектрической регистрацией спектра.
В процентах
|
Определяемый элемент |
Диапазон массовых долей элемента |
Определяемый элемент |
Диапазон массовых долей элемента |
|
Мышьяк |
0,0001 - 0,100 |
Хром |
0,0005 - 0,10 |
|
Сурьма |
0,0003 - 0,100 |
Кремний |
0,00030 - 0,0100 |
|
Свинец |
0,0001 - 0,100 |
Железо |
0,0003 - 0,100 |
|
Олово |
0,0001 - 0,10 |
Серебро |
0,0003 - 0,0100 |
|
Висмут |
0,00003 - 0,010 |
Фосфор |
0,0010 - 0,100 |
|
Цинк |
0,0003 - 0,0100 |
Кадмий |
0,00003 - 0,0010 |
|
Магний |
0,0002 - 0,0100 |
Кобальт |
0,00003 - 0,0010 |
|
Марганец |
0,00003 - 0,0100 |
Селен |
0,00003 - 0,00100 |
|
Никель |
0,0002 - 0,100 |
Теллур |
0,00005 - 0,0010 |
19.2 Требования к погрешности анализа
Погрешность результатов анализа массовых долей определяемых элементов, значения пределов повторяемости и воспроизводимости для доверительной вероятности Р = 0,95 должны соответствовать приведенным в таблице 31.
В процентах
|
Определяемый элемент |
Диапазон массовых долей элемента |
Погрешность результатов анализа ± Δ |
Предел |
|
|
повторяемости r (n=2) |
воспроизводимости R |
|||
|
Мышьяк |
От 0,00010 до 0,00030 включ. |
0,00006 |
0,00007 |
0,00008 |
|
Св. 0,0003 » 0,0010 » |
0,0001 |
0,0001 |
0,0002 |
|
|
» 0,0010 » 0,0030 » |
0,0003 |
0,0003 |
0,0007 |
|
|
» 0,0030 » 0,0100 » |
0,0008 |
0,0006 |
0,0012 |
|
|
» 0,010 » 0,030 » |
0,003 |
0,003 |
0,007 |
|
|
» 0,030 » 0,100 » |
0,015 |
0,010 |
0,025 |
|
|
Сурьма |
От 0,00030 до 0,00100 включ, |
0,00015 |
0,00014 |
0,00020 |
|
Св. 0,0010 » 0,0030 » |
0,0004 |
0,0003 |
0,0005 |
|
|
» 0,003 » 0,010 » |
0,001 |
0,001 |
0,002 |
|
|
» 0,010 » 0,030 » |
0,004 |
0,003 |
0,005 |
|
|
» 0,030 » 0,100 » |
0,008 |
0,007 |
0,012 |
|
|
Свинец |
От 0,00010 до 0,00030 включ. |
0,00006 |
0,00006 |
0,00008 |
|
Св. 0,00030 » 0,00100 » |
0,00015 |
0,00011 |
0,00021 |
|
|
» 0,0010 » 0,0030 » |
0,0004 |
0,0003 |
0,0005 |
|
|
» 0,0030 » 0,0100 » |
0,0014 |
0,0010 |
0,0020 |
|
|
» 0,010 » 0,030 » |
0,004 |
0,003 |
0,006 |
|
|
» 0,030 » 0,100 » |
0,014 |
0,011 |
0,020 |
|
|
Висмут |
От 0,00003 до 0,00010 включ. |
0,00002 |
0,00002 |
0,00003 |
|
Св. 0,00010 » 0,00030 » |
0,00005 |
0,00003 |
0,00007 |
|
|
» 0,0003 » 0,0010 » |
0,0002 |
0,0002 |
0,0003 |
|
|
» 0,0010 » 0,0030 » |
0,0006 |
0,0006 |
0,0008 |
|
|
» 0,003 » 0,010 » |
0,001 |
0,001 |
0,002 |
|
|
Олово |
От 0,00010 до 0,00030 включ. |
0,00007 |
0,00008 |
0,00010 |
|
Св. 0,0003 » 0,0010 » |
0,0002 |
0,0002 |
0,0003 |
|
|
» 0,0010 » 0,0030 » |
0,0007 |
0,0005 |
0,0009 |
|
|
» 0,0030 » 0,0100 » |
0,0018 |
0,0018 |
0,0025 |
|
|
» 0,010 » 0,030 » |
0,006 |
0,006 |
0,008 |
|
|
» 0,03 » 0,10 » |
0,01 |
0,01 |
0,02 |
|
|
Цинк |
От 0,0003 до 0,0010 включ. |
0,0002 |
0,0002 |
0,0003 |
|
Св. 0,0010 » 0,0030 » |
0,0006 |
0,0005 |
0,0008 |
|
|
» 0,0030 » 0,0100 » |
0,0018 |
0,0020 |
0,0025 |
|
|
Кремний |
От 0,00030 до 0,00060 включ. |
0,00013 |
0,00012 |
0,00018 |
|
Св. 0,00060 » 0,00100 » |
0,00019 |
0,00019 |
0,00027 |
|
|
» 0,0010 » 0,0030 » |
0,0004 |
0,0004 |
0,0006 |
|
|
» 0,0030 » 0,0100 » |
0,0014 |
0,0010 |
0,0020 |
|
|
Никель |
От 0,0002 до 0,0005 включ. |
0,0002 |
0,0002 |
0,0003 |
|
Св. 0,0005 » 0,0010 » |
0,0004 |
0,0004 |
0,0005 |
|
|
» 0,0010 » 0,0030 » |
0,0006 |
0,0006 |
0,0009 |
|
|
» 0,0030 » 0,0100 » |
0,0018 |
0,0020 |
0,0024 |
|
|
» 0,010 » 0,030 » |
0,003 |
0,003 |
0,004 |
|
|
» 0,030 » 0,100 » |
0,018 |
0,020 |
0,025 |
|
|
Магний |
От 0,00010 до 0,00030 включ. |
0,00007 |
0,00008 |
0,00008 |
|
Св. 0,0003 » 0,0010 » |
0,0002 |
0,0002 |
0,0003 |
|
|
» 0,0010 » 0,0030 » |
0,0006 |
0,0007 |
0,0009 |
|
|
» 0,0030 » 0,0100 » |
0,0014 |
0,0018 |
0,0020 |
|
|
Марганец |
От 0,00003 до 0,00010 включ. |
0,00002 |
0,00002 |
0,00003 |
|
Св. 0,00010 » 0,00030 » |
0,00006 |
0,00007 |
0,00008 |
|
|
» 0,0003 » 0,0010 » |
0,0002 |
0,0002 |
0,0003 |
|
|
» 0,0010 » 0,0030 » |
0,0006 |
0,0006 |
0,0009 |
|
|
» 0,0030 » 0,0100 » |
0,0018 |
0,0017 |
0,0025 |
|
|
Хром |
От 0,0005 до 0,0010 включ. |
0,0002 |
0,0003 |
0,0003 |
|
Св. 0,0010 » 0,0030 » |
0,0008 |
0,0008 |
0,0010 |
|
|
» 0,003 » 0,010 » |
0,002 |
0,002 |
0,003 |
|
|
» 0,010 » 0,030 » |
0,008 |
0,008 |
0,010 |
|
|
» 0,03 » 0,10 » |
0,02 |
0,02 |
0,03 |
|
|
Серебро |
От 0,0003 до 0,0010 включ. |
0,0002 |
0,0002 |
0,0003 |
|
Св. 0,0010 » 0,0030 » |
0,0008 |
0,0008 |
0,0010 |
|
|
» 0,0030 » 0,0100 » |
0,0014 |
0,0015 |
0,0020 |
|
|
Кадмий |
От 0,00003 до 0,00005 включ. |
0,00002 |
0,00002 |
0,00003 |
|
Св. 0,00005 » 0,00010 » |
0,00003 |
0,00003 |
0,00004 |
|
|
» 0,00010 » 0,00030 » |
0,00008 |
0,00008 |
0,00010 |
|
|
» 0,0003 » 0,0010 » |
0,0001 |
0,0001 |
0,0002 |
|
|
Фосфор |
От 0,0010 до 0,0030 включ. |
0,0008 |
0,0008 |
0,0010 |
|
Св. 0,003 » 0,010 » |
0,002 |
0,002 |
0,003 |
|
|
» 0,010 » 0,030 » |
0,006 |
0,007 |
0,008 |
|
|
» 0,030 » 0,100 » |
0,014 |
0,010 |
0,020 |
|
|
Теллур |
От 0,00005 до 0,00010 включ. |
0,00004 |
0,00004 |
0,00005 |
|
Св. 0,00010 » 0,00030 » |
0,00006 |
0,00007 |
0,00008 |
|
|
» 0,0003 » 0,0010 » |
0,0001 |
0,0001 |
0,0002 |
|
|
Кобальт |
От 0,00003 до 0,00010 включ. |
0,00002 |
0,00002 |
0,00003 |
|
Св. 0,00010 » 0,00030 » |
0,00005 |
0,00005 |
0,00007 |
|
|
» 0,0003 » 0,0010 » |
0,0001 |
0,0001 |
0,0002 |
|
|
Железо |
От 0,0003 до 0,0008 включ. |
0,0004 |
0,0003 |
0,0005 |
|
Св. 0,0006 » 0,0010 » |
0,0004 |
0,0005 |
0,0006 |
|
|
» 0,0010 » 0,0030 » |
0,0008 |
0,0008 |
0,0010 |
|
|
» 0,0030 » 0,0100 » |
0,0010 |
0,0010 |
0,0015 |
|
|
» 0,010 » 0,030 » |
0,004 |
0,003 |
0,005 |
|
|
» 0,030 » 0,100 » |
0,018 |
0,015 |
0,025 |
|
|
Селен |
От 0,00003 до 0,00010 включ. |
0,00002 |
0,00002 |
0,00003 |
|
Св. 0,00010 » 0,00030 » |
0,00006 |
0,00006 |
0,00008 |
|
|
» 0,00030 » 0,00100 » |
0,00018 |
0,00015 |
0,00025 |
|
19.3 Средства измерений, вспомогательные устройства, материалы, растворы
При выполнении анализа применяют следующие средства измерений, вспомогательные устройства:
- спектрометр дифракционный типа МФС-8;
- источник постоянного тока для питания дуги, обеспечивающий напряжение от 200 до 400 В и силу тока до 10 А;
- устройство для высокочастотного поджигания дуги постоянного тока от генератора любой системы (ПС-39,ДГ,ИГ);
- пресс гидравлический, обеспечивающий усилие от 1 до 10 МПа;
- пресс-форму из легированной стали (например, ХВГ), пуансон диаметром от 4 до 6 мм, высотой от 50 до 80 мм;
- печь муфельную с терморегулятором, позволяющую получать и поддерживать температуру не ниже 800°С;
- чашки платиновые по ГОСТ 6563 или фарфоровые по ГОСТ 9147, или кварцевые по ГОСТ 19908 выпарительные для растворения и выпаривания проб (для растворения можно применять также колбы или стаканы из термически стойкого стекла по ГОСТ 25336);
- электроды-подставки графитовые из угля марки ОСЧ, тип - кристаллический; марки, например ЭУЗ-М или ЭУЗ-П по ГОСТ 17022, диаметром от 6 до 10 мм. Для помещения брикетов или окисленных в кислороде таблеток на электродах-подставках высверливают углубления диаметром 6 мм и глубиной от 1,5 до 2 мм (рисунок 3);
а - расположение электродов и брикета до экспонирования; б - съемка в анодном режиме; в - съемка в катодном режиме 1 - графитовая подставка; 2 - брикет; 3 - подставной электрод; 4 - расплав
Рисунок 3 - Расположение электродов и брикетов
- электроды из меди марки М00 (или других марок с массовой долей меди не менее 99,97%) или из угля марки ОСЧ (С-2, С-3) в виде прутков диаметром от 6 до 7 мм, заточенные на полусферу или усеченный конус с площадкой диаметром от 1,5 до 1,7 мм;
- приспособление для заточки угольных или медных электродов, например станок модели КП-35;
- камеру кислородную для окисления СО и проб;
- баллон с кислородом, снабженный редуктором;
- шкаф сушильный лабораторный;
- электроплитку или баню песчаную;
- весы лабораторные специального класса точности по ГОСТ 24104;
- ступку агатовую или из органического стекла (допускается использование ступок фарфоровых по ГОСТ 9147);
- бюксы по ГОСТ 25336 для хранения брикетов или оксидных таблеток;
- пинцеты для захватывания таблеток или брикетов;
- колпачки стеклянные или пластмассовые для защиты от пыли заточенных электродов;
- магнит типа МВМ-63;
- секундомер по [28].
При выполнении анализа применяют следующие материалы, растворы:
- вату гигроскопическую по ГОСТ 5556; ГОСТ 31382-2009
- стандартные образцы состава меди или оксида меди или синтетические смеси;
- кислоту азотную особой чистоты по ГОСТ 11125 или кислоту азотную по ГОСТ 4461 (перегнанную), разбавленную 1:1 или 1:10;
- спирт этиловый по ГОСТ 18300. Расход спирта на одно определение-10 г;
- воду дистиллированную по ГОСТ 6709.
19.4 Метод анализа
Метод основан на измерении интенсивности спектральных линий определяемых элементов в анализируемом образце.
Пробы подвергают предварительному окислению плавлением на катоде дуги постоянного тока в атмосфере кислорода. Допускается превращение проб в оксиды растворением их в азотной кислоте, упариванием и прокаливанием.
Окисленный образец помещают на графитовую подставку и между ним и подставным электродом из чистой меди (или угля) возбуждают дугу постоянного тока с последующей фотоэлектрической регистрацией спектра. Метод дает возможность проводить анализ образца в любом виде.
19.5 Подготовка к выполнению анализа
19.5.1 Подготовка прибора к измерению
Подготовку прибора к выполнению измерений проводят в соответствии с требованиями действующей инструкции по эксплуатации спектрометра.
Спектрометр градуируют при создании метода с использованием СО состава меди и строят зависимость интенсивности аналитической линии от массовой доли для каждого определяемого элемента. При дальнейшей работе выполняют корректировку градуировочных характеристик в соответствии с инструкцией по эксплуатации спектрометра.
19.5.2 Пробу и стандартный образец в виде таблетки массой (0,50 ±0,05) г, диаметром 6 мм и высотой не более 2 мм изготовляют на любом металлорежущем оборудовании или вручную из любых кусков произвольной формы.
Пробы и СО необходимой массы могут быть отрезаны (отпилены) от стержней или спрессованы из стружки. Стружку предварительно отмагничивают. Затем стружку и СО в виде таблеток очищают от поверхностных загрязнений травлением в азотной кислоте, разбавленной 1:10. Стружку и таблетки СО промывают в дистиллированной воде, спирте и сушат. При прессовании таблеток из стружки матрицу и пуансон тщательно очищают от остатков ранее прессованной пробы (промывают водой и протирают спиртом). Готовят не менее двух таблеток проб и СО каждого состава.
19.5.3 Проводят окисление СО и проб в кислородной камере (предварительно все детали кислородной камеры и графитовые подставки для проб и СО очищают от оксидов меди). Поворотный столик укрепляют в нижнем электрододержателе камеры. Во избежание взаимного загрязнения образцов на графитовые подставки поворотного столика помещают таблетки одного состава.
В верхнем держателе укрепляют подставной электрод из меди или угля диаметром от 6 до 7 мм, рабочий конец которого заточен на усеченный конус с углом при вершине 45° и площадкой диаметром от 1,5 до 1,7 мм. Межэлектродный промежуток устанавливают от 1,5 до 2 мм. Таблетка служит катодом дуги постоянного тока, при силе тока 6 А. Воздух из камеры вытесняют, пропуская сжатый кислород через камеру в течение 30 с. При окислении таблеток давление кислорода в камере поддерживают несколько выше атмосферного. Таблетка под действием дуги в течение от 20 до 30 с расплавляется и превращается в каплю расплавленных оксидов. Ток выключают и подводят к подставному электроду следующую таблетку.
19.5.4 Для измерения оксидных образцов от средней пробы отбирают две навески массой от 5,0 до 10,0 г. Навески помещают в выпарительные чашки. Приливают разбавленную 1:1 азотную кислоту из расчета 10 см3 на 1 г меди, растворяют при нагревании и выпаривают до сухих солей. Затем чашки помещают в муфельную печь и прокаливают при температуре (400 ±50)°С в течение 30 мин до прекращения выделения оксидов азота. Полученный порошок растирают в агатовой (или другой) ступке. Ступку и пестик предварительно протирают спиртом. Порошком наполняют кратеры угольных электродов или прессуют в таблетки (не менее 2). Масса навески пробы и СО должна быть одинаковой от (0,30 ±0,05) до (0,60 ±0,05) г.
19.5.5 Приготовление синтетических смесей приведено в Приложении А.
19.6 Выполнение анализа
19.6.1 Торцевую часть электродов для удаления поверхностных загрязнений прокаливают в дуге постоянного тока при величине тока от 6 до 10 А в течение 20 с, включая электрод-подставку в качестве анода дуги.
Подготовленные к измерению пробы и СО помещают на прокаленные графитовые подставки. В качестве подставного электрода применяют угли марки ОСЧ или медные стержни. Форма и размер электродов и их расположение во время экспозиции приведены на рисунке 3.
19.6.2 Для определения массовой доли мышьяка, сурьмы, свинца, олова, висмута, цинка, фосфора, кадмия, селена и теллура графитовую подставку с помещенной на нее пробой или СО используют в качестве анода дуги. При включении тока до расплавления образца дуга загорается между подставным электродом и подставкой, и после расплавления анодное пятно дуги переходит на образовавшийся расплав оксидов. Начало экспозиции отсчитывают после перехода анодного пятна дуги на образец. В течение всего времени экспозиции необходимо корректировать первоначально установленный дуговой промежуток по увеличенному изображению дуги на экране средней линзы осветительной системы или с использованием специальной короткофокусной проекционной линзы.
Регистрацию спектров проводят при ширине щели 0,035 мм, разрядном промежутке 3 мм, силе тока дуги от 6 до 10 А, времени экспозиции от 20 до 40 с.
19.6.3 Далее определяют массовые доли кобальта, магния, марганца, никеля, хрома, кремния, железа и серебра.
Регистрацию спектров проводят при разрядном промежутке 3 мм с применением дуги постоянного или переменного тока силой от 5 до 8 А на первом этапе и от 8 до 10 А на втором, время экспозиции - от 30 до 60 с в абсолютном или относительном режимах.
Начало экспозиции отсчитывают после перехода катодного пятна дуги на расплавленную часть королька.
Условия съемки спектрограммы - в соответствии с инструкцией по эксплуатации спектрометра.
19.6.4 В случаях, когда условия съемки спектров отличаются от рекомендуемых, следует предварительно подобрать оптимальные интервалы оптических плотностей линий в соответствии с инструкцией к прибору.
Длины волн аналитических линий для дифракционного спектрометра МФС-8 приведены в таблице 32.
Допускается применение других аналитических линий при условии, что они обеспечивают метрологические характеристики результатов анализа, приведенных в настоящем методе.
|
Определяемый элемент |
Длина волны аналитической линии, нм |
Определяемый элемент |
Длина волны аналитической линии, нм |
|
Висмут |
306,772 |
Олово |
286,332 |
|
Железо |
302,197 |
Свинец |
283,307 |
|
Кадмий |
214,441 |
Селен |
203,980 |
|
Кобальт |
345,351 |
Серебро |
338,289 |
|
Кремний |
251,611 |
Сурьма |
231,147 |
|
Магнии |
277,983 |
Теллур |
238,325 |
|
Марганец |
279,480 |
Фосфор |
253,561 |
|
Мышьяк |
234,984 |
Хром |
357,868 |
|
Никель |
341,477 |
Цинк |
334,502 |
19.7 Обработка результатов анализа
19.7.1 Обработку результатов анализа проводит компьютер по заданной программе и представляет их в виде массовых долей определяемых элементов.
19.7.2 За результат анализа принимают среднеарифметическое значение двух параллельных определений при условии, что абсолютная разность между ними в условиях повторяемости при доверительной вероятности Р = 0,95 не превышает значений предела повторяемости r, приведенных в таблице 31.
Если расхождение между результатами параллельных определений превышает значение предела повторяемости, выполняют процедуры, изложенные в стандарте [2] (подпункт 5.2.2.1).
19.8 Контроль точности результатов анализа
Контроль точности результатов анализа - в соответствии с 3.14.
19.9 Оформление результатов анализа
Результаты анализа оформляют в соответствии с 3.15, значения погрешности результатов анализа А приведены в таблице 31.
20 Метод эмиссионно-спектрального анализа с фотоэлектрической регистрацией спектра
20.1 Область применения
В настоящем разделе установлено определение массовых долей примесей в меди марок М00к и М00 в диапазонах, указанных в таблице 33, эмиссионно-спектральным методом с фотоэлектрической регистрацией спектра.
В процентах
|
Определяемый элемент |
Диапазон массовых долей элемента |
Определяемый элемент |
Диапазон массовых долей элемента |
|
Висмут |
0,000008 - 0,00100 |
Свинец |
0,000005 - 0,00070 |
|
Железо |
0,00007 - 0,0030 |
Селен |
0,00008 - 0,00200 |
|
Кадмий |
0,000030 - 0,00050 |
Серебро |
0,00008 - 0,00250 |
|
Кобальт |
0,000050 - 0,00100 |
Сурьма |
0,00007 - 0,00200 |
|
Кремний |
0,00009 - 0,00200 |
Теллур |
0,00005 - 0,00100 |
|
Марганец |
0,000010 - 0,00050 |
Фосфор |
0,0002 - 0,00200 |
|
Мышьяк |
0,000030 - 0,00200 |
Хром |
0,00002 - 0,00050 |
|
Никель |
0,00007 - 0,00200 |
Цинк |
0,00005 - 0,00200 |
|
Олово |
0,000020 - 0,00200 |
|
|
20.2 Общие требования
20.2.1 Общие требования к методу анализа, средствам измерений, вспомогательным устройствам, материалам и требования безопасности при выполнении анализа - в соответствии с разделами 3 и 4.
20.2.2 Массовую долю примесей определяют не менее чем по двум навескам меди.
20.3 Требования к погрешности анализа
Погрешность результатов анализа массовых долей определяемых элементов, значения пределов повторяемости и воспроизводимости для доверительной вероятности Р = 0,95 должны соответствовать приведенным в таблице 34.
В процентах
|
Наименование элемента |
Диапазон массовых долей элемента |
Погрешность результатов анализа ±Δ |
Предел |
|
|
повторяемости r (n=2) |
воспроизводимости R |
|||
|
Висмут |
От 0,000008 до 0,000020 включ. |
0,000005 |
0,000005 |
0,000006 |
|
Св. 0,000020 » 0,000060 » |
0,000009 |
0,000010 |
0,000013 |
|
|
» 0,000060 » 0,000150 » |
0,000025 |
0,000029 |
0,000031 |
|
|
» 0,00015 » 0,00040 » |
0,00004 |
0,00005 |
0,00006 |
|
|
» 0,00040 » 0,00100 » |
0,00008 |
0,00007 |
0,00011 |
|
|
Железо |
От 0,00007 до 0,00020 включ. |
0,00004 |
0,00005 |
0,00006 |
|
Св. 0,00020 » 0,00060 » |
0,00010 |
0,00012 |
0,00014 |
|
|
» 0,00060 » 0,00100 » |
0,00023 |
0,00030 |
0,00033 |
|
|
» 0,0010 » 0,0030 » |
0,0004 |
0,0006 |
0,0007 |
|
|
Кадмий |
От 0,000030 до 0,000090 включ. |
0,000015 |
0,000013 |
0,000019 |
|
Св. 0,000090 » 0,000200 » |
0,000029 |
0,000025 |
0,000036 |
|
|
» 0,00020 » 0,00050 » |
0,00005 |
0,00006 |
0,00007 |
|
|
Кобальт |
От 0,000050 до 0,000100 включ. |
0,000021 |
0,000014 |
0,000026 |
|
Св. 0,00010 » 0,00030 » |
0,00004 |
0,00003 |
0,00005 |
|
|
» 0,00030 » 0,00060 » |
0,00006 |
0,00005 |
0,00007 |
|
|
» 0,00060 » 0,00100 » |
0,00007 |
0,00007 |
0,00010 |
|
|
Кремний |
От 0,00009 до 0,00025 включ. |
0,00007 |
0,00007 |
0,00009 |
|
Св. 0,00025 » 0,00070 » |
0,00010 |
0,00010 |
0,00014 |
|
|
» 0,00070 » 0,00200 » |
0,00022 |
0,00025 |
0,00029 |
|
|
Мышьяк |
От 0,000030 до 0,000080 включ. |
0,000024 |
0,000020 |
0,000030 |
|
Св. 0,00008 » 0,00020 » |
0,00005 |
0,00004 |
0,00006 |
|
|
» 0,00020 » 0,00060 » |
0,00006 |
0,00009 |
0,00010 |
|
|
» 0,00060 » 0,00120 » |
0,00008 |
0,00010 |
0,00011 |
|
|
» 0,00120 » 0,00200 » |
0,00020 |
0,00024 |
0,00025 |
|
|
Никель |
От 0,00007 до 0,00020 включ. |
0,00005 |
0,00005 |
0,00007 |
|
Св. 0,00020 » 0,00060 » |
0,00006 |
0,00008 |
0,00009 |
|
|
» 0,00060 » 0,00100 » |
0,00008 |
0,00011 |
0,00012 |
|
|
» 0,00100 » 0,00200 » |
0,00014 |
0,00020 |
0,00021 |
|
|
Олово |
От 0,000020 до 0,000050 включ. |
0,000010 |
0,000013 |
0,000014 |
|
Св. 0,000050 » 0,000150 » |
0,000028 |
0,000037 |
0,000039 |
|
|
» 0,00015 » 0,00040 » |
0,00006 |
0,00006 |
0,00007 |
|
|
» 0,00040 » 0,00100 » |
0,00008 |
0,00008 |
0,00012 |
|
|
» 0,00100 » 0,00200 » |
0,00018 |
0,00020 |
0,00023 |
|
|
Свинец |
От 0,000005 до 0,000015 включ. |
0,000003 |
0,000004 |
0,000005 |
|
Св. 0,000015 » 0,000040 » |
0,000010 |
0,000010 |
0,000013 |
|
|
» 0,000040 » 0,000100 » |
0,000024 |
0,000023 |
0,000030 |
|
|
» 0,00010 » 0,00030 » |
0,00004 |
0,00005 |
0,00006 |
|
|
» 0,00030 » 0,00070 » |
0,00007 |
0,00008 |
0,00010 |
|
|
Серебро |
От 0,00008 до 0,00020 включ. |
0,00004 |
0,00005 |
0,00006 |
|
Св. 0,00020 » 0,00050 » |
0,00010 |
0,00009 |
0,00012 |
|
|
» 0,00050 » 0,00150 » |
0,00012 |
0,00013 |
0,00017 |
|
|
» 0,00150 » 0,00250 » |
0,00029 |
0,00025 |
0,00036 |
|
|
Сурьма |
От 0,00007 до 0,00020 включ. |
0,00005 |
0,00003 |
0,00007 |
|
Св. 0,00020 » 0,00060 » |
0,00010 |
0,00008 |
0,00013 |
|
|
» 0,00060 » 0,00100 » |
0,00019 |
0,00010 |
0,00024 |
|
|
» 0,00100 » 0,00200 » |
0,00022 |
0,00020 |
0,00029 |
|
|
Селен |
От 0,00008 до 0,00020 включ. |
0,00005 |
0,00003 |
0,00007 |
|
Св. 0,00020 » 0,00050 » |
0,00010 |
0,00008 |
0,00013 |
|
|
» 0,00050 » 0,00100 » |
0,00019 |
0,00010 |
0,00024 |
|
|
» 0,0010 » 0,0020 » |
0,00022 |
0,00020 |
0,00029 |
|
|
Теллур |
От 0,00005 до 0,00015 включ. |
0,00004 |
0,00004 |
0,00005 |
|
Св. 0,00015 » 0,00040 » |
0,00006 |
0,00007 |
0,00008 |
|
|
» 0,00040 » 0,00100 » |
0,00010 |
0,00011 |
0,00013 |
|
|
Фосфор |
От 0,00020 до 0,00060 включ. |
0,00010 |
0,00010 |
0,00014 |
|
Св. 0,00060 » 0,00100 » |
0,00012 |
0,00014 |
0,00017 |
|
|
» 0,00100 » 0,00200 » |
0,00015 |
0,00020 |
0,00021 |
|
|
Хром |
От 0,000020 до 0,000060 включ. |
0,000016 |
0,000020 |
0,000021 |
|
Св. 0,00006 » 0,00010 » |
0,00004 |
0,00003 |
0,00005 |
|
|
» 0,00010 » 0,00030 » |
0,00005 |
0,00007 |
0,00008 |
|
|
» 0,00030 » 0,00050 » |
0,00006 |
0,00009 |
0,00010 |
|
|
Цинк |
От 0,00005 до 0,00010 включ. |
0,00004 |
0,00003 |
0,00005 |
|
Св. 0,00010 » 0,00030 » |
0,00006 |
0,00005 |
0,00008 |
|
|
» 0,00030 » 0,00070 » |
0,00009 |
0,00008 |
0,00013 |
|
|
» 0,00070 » 0,00200 » |
0,00015 |
0,00014 |
0,00021 |
|
|
Марганец |
От 0,000010 до 0,000030 включ. |
0,000008 |
0,000008 |
0,000010 |
|
Св. 0,000030 » 0,000090 » |
0,000017 |
0,000015 |
0,000022 |
|
|
» 0,000090 » 0,000200 » |
0,000024 |
0,000024 |
0,000034 |
|
|
» 0,00020 » 0,00050 » |
0,00003 |
0,00004 |
0,00005 |
|
20.4 Средства измерений, вспомогательные устройства, материалы, растворы
При выполнении анализа применяют следующие средства измерений, вспомогательные устройства:
- спектрометр дифракционный типа МФС-8;
- генератор УГЭ-4 или другой источник постоянного тока с устройством для высокочастотного поджигания дуги, реостатом и амперметром, обеспечивающим напряжение от 200 до 400 В и силу тока до 12 А;
- электрошкаф сушильный лабораторный;
- электропечь сопротивления камерную лабораторную с температурой нагрева до 900°С;
- станок для заточки графитовых электродов, например модель КП-35 или другого типа;
- пресс-форму из легированной стали (например, ХВГ), пуансон диаметром от 4 до 6 мм, высотой от 50 до 80 мм;
- твердомер ТШ-2 или пресс, обеспечивающий получение усилия от 9,8 до 14,7 кН;
- весы лабораторные высокого класса точности по ГОСТ 24104;
- секундомеры механические по [28];
- ступку агатовую с пестиком или чашу фарфоровую глазурованную с медным пестиком;
- чаши из кварцевого стекла по ГОСТ 19908 или чаши фарфоровые по ГОСТ 9147;
- электроды-подставки графитовые, электрододержатели медные, охлаждаемые водой, в соответствии с рисунком 4, обеспечивающие теплоотвод для предотвращения полного расплава королька при силе тока 10 А;
- установку для перегонки азотной кислоты;
- установку для двойной перегонки воды типа ВС или другого типа;
- деионизатор типа Barnstead E-pure или любой другой;
- магнит;
- пинцет медицинский;
- стаканчики для взвешивания по ГОСТ 25336;
- банки пластмассовые с крышками;
1 - штуцер для входа охлаждающей воды; 2 - штуцер для выхода охлаждающей воды
Рисунок 4 - Электрододержатели
- электроды графитовые из спектральных углей марки ОСЧ или С-3 в виде прутков (в соответствии с рисунком 5) со штифтообразной заточкой с площадкой диаметром 3 мм.
а - до экспонирования; б - при анодной полярности таблетки; в - при катодной полярности таблетки
1 - графитовый электрод-подставка; 2 - таблетка пробы или стандартного образца; 3 - графитовый электрод; 4 - капля расплава
Рисунок 5 - Графитовые электроды
При выполнении анализа применяют следующие материалы, растворы:
- вату медицинскую гигроскопическую по ГОСТ 5556;
- бумагу масштабно-координатную марки ЛН по ГОСТ 334;
- воду дистиллированную по ГОСТ 6709;
- воду деионизированную, полученную пропусканием дистиллированной воды через ионообменную колонку с катионитом (например, марки КУ-2-8), или воду бидистиллированную;
- катионит марки КУ-2-8 по ГОСТ 20298;
- кислоту азотную особой чистоты по ГОСТ 11125 или кислоту азотную по ГОСТ 4461 (перегнанную в установке), разбавленную 1:1, 2:1;
- спирт этиловый по ГОСТ 18300;
- СО состава меди.
20.5 Метод анализа
Метод основан на измерении интенсивностей спектральных линий определяемых элементов спектрометром. Спектр излучения возбуждается дугой постоянного тока между подставным графитовым электродом и таблеткой оксида меди, полученной путем растворения пробы в азотной кислоте и термического разложения образовавшихся солей.
20.6 Подготовка к выполнению анализа
20.6.1 Подготовка прибора
Подготовку прибора к выполнению измерений проводят в соответствии с требованиями действующей инструкции по эксплуатации спектрометра.
Спектрометр градуируют при создании метода с использованием СО состава меди и строят зависимость интенсивности аналитической линии от массовой доли для каждого определяемого элемента. При дальнейшей работе выполняют корректировку градуировочных характеристик в соответствии с инструкцией по эксплуатации спектрометра.
20.6.2 Приготовление синтетических смесей, которые могут использоваться в качестве СО для градуировки, приведено в Приложении А.
20.6.3 Перед выполнением измерений ступку, пресс-форму, чаши и пинцет протирают спиртом. Расход спирта на одно определение - 10 см3. После сжигания каждой пробы электрододержатели также протирают спиртом.
От пробы отбирают две навески массой от 5 до 10 г. Навески помещают в выпаривательные чаши, приливают азотную кислоту, разбавленную 1:1, из расчета от 8 до 10 см3 кислоты на 1 г пробы, и растворяют при нагревании.
Полученные растворы выпаривают досуха, чаши с сухими солями помещают в печь и прокаливают при температуре (500 ±25)°С в течение 30 мин с момента прекращения выделения паров оксида азота. Полученный оксид меди растирают в ступке. Отбирают по три навески массой 0,500 г от порошков оксида меди, полученных из каждой исходной навески пробы, и прессуют таблетки (предварительно пресс-форма должна быть очищена и протерта спиртом).
Допускается изменение массы порошка для изготовления таблеток от 0,500 до 1,000 г с целью достижения нижних границ диапазона массовых долей определяемых элементов.
Образцы для градуировки в виде оксидных порошков также прессуют в таблетки. Массы порошка при изготовлении таблеток для градуировки и для измерения массовой доли примеси в анализируемой пробе должны быть одинаковыми.
Подготовленные к анализу таблетки хранят в бюксах, пластмассовых или полиэтиленовых банках с завинчивающимися крышками.
20.6.4 Построение градуировочных графиков
Для построения градуировочных графиков и установления градуировочных зависимостей используют синтетические смеси, приготовленные согласно приложению А, или СО состава меди любой категории. Количество образцов - не менее четырех, массовые доли определяемых элементов в СО состава должны охватывать пределы диапазона измерений.
По 20.7.1 и 20.7.2 выполняют измерения трех таблеток каждого образца и оксида меди (для учета фона), приготовленного в соответствии с приложением А. По полученным значениям интенсивностей спектральных линий и соответствующим им значениям массовых долей элементов строят градуировочные графики в прямоугольных координатах, используя для каждой точки среднее значение из двух измерений интенсивностей спектральных линий.
20.7 Выполнение анализа
20.7.1 Таблетки проб помещают на графитовые электроды-подставки. Торцевую часть электродов для удаления поверхностных загрязнений прокаливают в дуге постоянного тока в течение 20 с при силе тока от 6 до 10 А, используя электрод-подставку в качестве анода дуги в соответствии с рисунком 5. Для верхних электродов применяют графитовые стержни марки ОСЧ или предварительно обожженные стержни марки С-3.
20.7.2 Измерения проводят в два этапа: первый - при анодной и второй - при катодной полярности таблетки.
20.7.2.1 На первом этапе определяют массовые доли легколетучих примесей: кадмия, селена, мышьяка, теллура, свинца, висмута, фосфора, сурьмы, цинка, олова. Предварительно проводят обжиг таблетки, используя графитовую подставку с помещенной на нее таблеткой в качестве анода. Для этого сначала зажигают дугу постоянного тока между верхним электродом и подставкой, а после сплавления части таблетки анодное пятно дуги переходит на образовавшийся расплав (королек) оксидов. Продолжительность обжига с момента перехода дуги на расплав - 5 с.
По окончании обжига начинают аналитическую экспозицию продолжительностью 40 с в абсолютном режиме.
Первоначально установленный межэлектродный промежуток периодически корректируют в течение всей экспозиции по увеличенному изображению дуги на экране специальной короткофокусной проекционной системы, перемещая электрододержатели.
Регистрацию спектров проводят при ширине входной щели 0,035 мм, освещении входной щели растровым конденсором, разрядном промежутке 30 мм, силе тока дуги от 5 до 8 А, времени экспозиции от 20 до 40 с.
20.7.2.2 На втором этапе определяют массовые доли труднолетучих примесей: железа, марганца, никеля, серебра, кобальта, кремния, хрома.
Образующийся на первом этапе анализа королек помещают на свежезаточенную графитовую подставку и проводят его обжиг в течение 5 с, используя графитовую подставку в качестве катода.
Регистрацию спектров проводят при разрядном промежутке 3 мм, силе тока дуги от 5 до 8 Δ, времени экспозиции от 20 до 30 с в абсолютном или относительном режимах.
20.7.3 Для каждой таблетки регистрируют показания выходного измерительного прибора, пропорциональные интенсивности спектральной линии определяемого элемента при длине волны, приведенной в таблице 35.
|
Определяемый элемент |
Длина волны, нм |
Определяемый элемент |
Длина волны, нм |
|
Висмут |
306,772 |
Свинец |
283,307 |
|
Железо |
302,107 |
Селен |
203,980 |
|
Кадмий |
214,441 |
Серебро |
338,289 |
|
Кобальт |
345,351 |
Сурьма |
231,147 |
|
Кремний |
251,611 |
Теллур |
238,325 |
|
Марганец |
279,480 |
Фосфор |
253,561 |
|
Мышьяк |
234,984 |
Хром |
357,868 |
|
Никель |
341,477 |
Цинк |
334,502 |
|
Олово |
286,332 |
|
|
|
Примечания 1 Допускается применение других условий проведения анализа и других аналитических линий, обеспечивающих получение метрологических характеристик в соответствии с требованиями настоящего метода 2 Допускается определение магния в анализируемой пробе. |
|||
20.8 Обработка результатов анализа
20.8.1 Обработку результатов анализа проводит компьютер по заданной программе и представляет их в виде массовых долей определяемых элементов.
20.8.2 За результат анализа принимается среднеарифметическое значение двух параллельных определений при условии, что абсолютная разность между ними в условиях повторяемости не превышает значений (при доверительной вероятности Р = 0,95) предела повторяемости r, приведенных в таблице 34.
Если расхождение между результатами параллельных определений превышает значение предела повторяемости, выполняют процедуры, изложенные в стандарте [2] (подпункт 5.2.2.1).
20.9 Контроль точности результатов анализа
Контроль точности результатов анализа - в соответствии с 3.14.
20.10 Оформление результатов анализа
Результаты анализа оформляют в соответствии с 3.15, значения погрешности результатов анализа А приведены в таблице 34.
Приложение
А
(рекомендуемое)
Приготовление синтетических смесей
Синтетические смеси представляют собой порошки оксида меди, полученные путем растворения чистой основы в азотной кислоте, введения дозируемых добавок растворов примесей, последующего выпаривания и термического разложения смеси нитратов. Синтетические смеси могут использоваться в качестве стандартных образцов для градуировки.
А.1 Средства измерений, вспомогательные устройства, материалы, растворы
При приготовлении синтетических смесей применяют следующие средства измерений, вспомогательные устройства:
- весы лабораторные специального класса точности по ГОСТ 24104;
- электропечь камерную любого типа с терморегулятором;
- стаканы В-1-50 ТХС, В-1- 250 ТХС, В-1-2000 ТХС по ГОСТ 25336;
- колбы конические Кн-2-250-34 ТХС по ГОСТ 25336;
- колбы мерные 2-100-2, 2-250-2 по ГОСТ 1770;
- чаши, ступки фарфоровые по ГОСТ 9147 или ступку агатовую или яшмовую;
- пипетки 1-2-1-5, 1-2-1-10, 1-2-1-25 по ГОСТ 29227;
- чаши кварцевые по ГОСТ 19908;
- бюретку 1-1-2-25-0,1 по ГОСТ 29251;
- банки полиэтиленовые, фторопластовые с завинчивающимися крышками или бюксы по ГОСТ 25336.
При приготовлении синтетических смесей применяют следующие материалы, растворы:
- железо карбонильное особо чистое по [19];
- висмут марки Ви00 по ГОСТ 10928;
- медь марки М00к по ГОСТ 859;
- олово не ниже марки 01 по ГОСТ 860;
- кадмий марки Кд0 по ГОСТ 1467;
- никель марки Н1у по ГОСТ 849;
- серебро по ГОСТ 6836;
- кобальт марки К0 по ГОСТ 123;
- хром марки Х99Н1 по ГОСТ 5905;
- марганец марки Мр00 по ГОСТ 6008;
- свинец марки С0 по ГОСТ 3778;
- фосфор по ГОСТ 8655;
- мышьяк металлический по [29];
- теллур особой чистоты по [27];
- селен по ГОСТ 10298;
- цинк марки Ц0 по ГОСТ 3640;
- сурьму марки Су00 по ГОСТ 1089;
- свинец (II) азотнокислый по ГОСТ 4236, перекристаллизованный, или свинец по ГОСТ 3778;
- натрий кремнекислый мета 9-водный (или тетраэтоксисилан);
- кислоту азотную по ГОСТ 4461 (перегнанную в кварцевом аппарате) или кислоту азотную особой чистоты по ГОСТ 11125, разбавленную 2:1, 1:1, 1:2;
- кислоту соляную по ГОСТ 3118;
- кислоту винную по ГОСТ 5817;
- кислоту щавелевую по ГОСТ 22180;
- натрия гидроксид (натрия гидроокись) по ГОСТ 4328, раствор массовой концентрации 100 г/дм3;
- спирт этиловый по ГОСТ 18300;
- серебро азотнокислое по ГОСТ 1277, раствор массовой концентрации 20 г/дм3;
- воду деионизированную, полученную пропусканием дистиллированной воды по ГОСТ 6709 через ионообменную колонку с катионитом или любым другим способом, или воду бидистиллированную, полученную перегонкой в кварцевом аппарате;
- магний по ГОСТ 804.
А.2 Приготовление растворов примесей известной концентрации
А.2.1 Каждую навеску массой 0,6250 г никеля, марганца, магния, кобальта, железа, цинка, свинца, висмута, кадмия, фосфора, серебра, селена, теллура растворяют в 25 см3 азотной кислоты в отдельных стаканах вместимостью 250 см3 каждый, растворы переводят в мерные колбы вместимостью 250 см3 и доливают до метки азотной кислотой, разбавленной 1:1.
1 см3 каждого раствора содержит 2,5 мг каждой из вышеуказанных примесей.
А.2.2 Навеску хрома массой 0,6250 г растворяют в объеме от 20 до 30 см3 соляной кислоты на кипящей водяной бане. Затем раствор упаривают до сухих солей. Добавляют от 5 до 10 см3 азотной кислоты и раствор упаривают до влажных солей. Операцию обработки солей азотной кислотой повторяют еще три раза, каждый раз выпаривая до влажных солей. Затем приливают 100 см3 азотной кислоты, разбавленной 1:1, помещают полученный раствор в мерную колбу вместимостью 250 см3 и доливают до метки азотной кислотой, разбавленной 1:1.
От полученного раствора отбирают от 10 до 20 см3 и помещают в стакан вместимостью 50 см3 для проверки на присутствие хлорид-иона с использованием раствора азотнокислого серебра. Если в растворе обнаружено присутствие хлорид-иона, операцию обработки солей азотной кислотой повторяют.
1 см3 раствора содержит 2,5 мг хрома.
А.2.3 Навеску олова массой 0,6250 г помещают в стакан вместимостью 250 см3, добавляют 5 г щавелевой кислоты, а затем 20 см3 азотной кислоты, разбавленной 2:1, и выдерживают без нагревания до растворения навески. Переносят раствор в мерную колбу вместимостью 250 см3 и доливают до метки азотной кислотой, разбавленной 2:1.
1 см3 раствора содержит 2,5 мг олова.
А.2.4 Навеску сурьмы массой 0,6250 г помещают в стакан вместимостью 250 см3, добавляют 4 г винной кислоты и затем растворяют в избытке горячей азотной кислоты при кипячении. Раствор переносят в мерную колбу вместимостью 250 см3 и доливают до метки азотной кислотой, разбавленной 1:1.
1 см3 раствора содержит 2,5 мг сурьмы.
А.2.5 Навеску кремнекислого натрия массой 1,0117 г помещают в мерную колбу вместимостью 100 см3, растворяют в объеме от 5 до 7 см3 воды и доливают до метки азотной кислотой, разбавленной 1:2, или навеску тетраэтоксисилана массой 0,7418 г помещают в мерную колбу вместимостью 100 см3, растворяют в этиловом спирте и доливают до метки спиртом.
1 см3 раствора содержит 1 мг кремния.
А.2.6 Навеску мышьяка, предварительно очищенного от оксидной пленки, массой 0,6250 г помещают в стакан вместимостью 250 см3, приливают от 125 до 150 см3 кипящей азотной кислоты и растворяют при нагревании. После охлаждения раствор переносят в мерную колбу вместимостью 250 см3 и доливают до метки азотной кислотой, разбавленной 1:1.
1 см3 раствора содержит 2,5 мг мышьяка.
А.2.7 Навеску азотнокислого свинца массой 0,9988 г помещают в мерную колбу вместимостью 250 см3, растворяют в объеме от 10 до 15 см3 воды и доливают до метки азотной кислотой, разбавленной 1:1.
1 см3 раствора содержит 2,5 мг свинца.
А.3 Приготовление синтетических смесей
А.3.1 Приготовление растворов 1 - 5
Раствор 1: в мерную колбу вместимостью 250 см3 помещают по 10 см3 азотнокислых растворов олова, кобальта, марганца, магния, висмута, кадмия, мышьяка, теллура, хрома и доливают до метки азотной кислотой, разбавленной 1:1.
1 см3 раствора 1 содержит по 0,1 мг указанных примесей.
Раствор 2: в мерную колбу вместимостью 250 см3 помещают по 10 см3 азотнокислых растворов свинца, никеля, сурьмы, цинка, селена и доливают до метки азотной кислотой, разбавленной 1:1.
1 см3 раствора 2 содержит по 0,1 мг указанных примесей.
Раствор 3: в мерную колбу вместимостью 100 см3 помещают по 20 см3 азотнокислых растворов фосфора, железа, серебра и доливают до метки азотной кислотой, разбавленной 1:1.
1 см3 раствора 3 содержит по 0,5 мг указанных примесей.
Раствор 4: 10 см3 раствора кремнекислого натрия помещают в мерную колбу вместимостью 100 см3 и доливают до метки азотной кислотой, разбавленной 1:2, или 10 см3 спиртового раствора тетраэтоксисилана помещают в мерную колбу вместимостью 100 см3 и доливают до метки спиртом.
1 см3 раствора 4 содержит 0,1 мг кремния.
Раствор 5: в мерную колбу вместимостью 250 см3 помещают по 1 см3 азотнокислых растворов висмута, свинца и марганца и доливают до метки азотной кислотой, разбавленной 1:1.
1 см3 раствора 5 содержит по 0,01 мг указанных примесей.
А.3.2 Приготовление растворов меди
В ряд стаканов (в зависимости от количества смесей) вместимостью 2000 см3 каждый помещают по 200 г меди, приливают азотную кислоту, разбавленную 1:1 (из расчета от 7 до 8 см3 на 1 г меди), и растворяют при нагревании.
А.3.3 Приготовление смеси оксидов
В полученные растворы меди вводят рассчитанные объемы (в зависимости от массовых долей определяемых элементов) растворов 1-5, выпаривают в кварцевых чашах до сухих солей, соли прокаливают в камерной печи при температуре от 400°С до 450°С до полного удаления оксидов азота.
Смеси оксидов растирают в ступке или измельчают любым способом, исключающим загрязнение материала синтетической смеси определяемыми элементами. Массовые доли элементов в синтетических смесях приведены в таблице А. 1.
Таблица А.1
|
Обозначение смесей |
Массовая доля элементов, % |
||||
|
Висмут, свинец, марганец |
Кобальт, олово, мышьяк, магний, кадмий, теллур, хром, железо, серебро |
Селен, никель, сурьма, цинк |
Фосфор |
Кремний |
|
|
СМИ |
0,000005 |
- |
- |
- |
- |
|
СМ-2 |
0,00001 |
0,00002 |
0,00005 |
0,0002 |
0,00005 |
|
СМ-3 |
0,00005 |
0,00005 |
0,0001 |
0,0005 |
0,0001 |
|
СМ-4 |
0,0001 |
0,0001 |
0,0005 |
0,001 |
0,0005 |
|
СМ-5 |
0,0002 |
0,0002 |
0,001 |
0,002 |
0,001 |
|
СМ-6 |
0,0005 |
0,0005 |
0,002 |
0,005 |
0,002 |
|
СМ-7 |
0,005 |
0,005 |
0,005 |
0,01 |
0,005 |
|
СМ-8 |
0,01 |
0,01 |
0,01 |
0,05 |
0,01 |
|
СМ-9 |
0,05 |
0,05 |
0,05 |
0,1 |
- |
|
СМ-10 |
0,1 |
0,1 |
0,1 |
- |
- |
Смеси и оксид меди без примесей хранят в бюксах или банках с завинчивающимися крышками. Способ хранения должен исключать возможность загрязнения и увлажнения смесей. При соблюдении этих условий срок хранения смесей - 5 лет.
Допускается изменение массы навесок меди и примесей (с соответствующим пересчетом) в зависимости от потребности в смесях, массовых долей примесей и состава анализируемых проб.
Библиография
|
[1] ИСО 5725-11:1994* |
Точность (правильность и прецизионность) методов и результатов измерений. Часть 1. Основные положения и определения |
|
|
[2] ИСО 5725-6:1994** |
Точность (правильность и прецизионность) методов и результатов измерений. Часть 6. Использование значений точности на практике |
|
|
[3] Рекомендации по межгосударственной стандартизации |
Государственная система обеспечения единства измерений. Внутренний контроль качества результатов количественного химического анализа |
|
|
[4] ГОСТ Р 50779.42-99 (ИСО 8258-91) |
Статистические методы. Контрольные карты Шухарта |
|
|
[5] Рекомендации по межгосударственной стандартизации |
Государственная система обеспечения единства измерений. Смеси аттестованные. Общие требования к разработке |
|
|
[6] Гигиенические нормативы |
Химические факторы производственной среды. Предельно допустимые концентрации (ПДК) вредных веществ в воздухе рабочей зоны |
|
|
Правила пожарной безопасности в Российской Федерации |
||
|
[8] ПБ 03-576-03*** |
Правила устройства и безопасной эксплуатации сосудов, работающих под давлением, утвержденные Постановлением Госгортехнадзора РФ от 11 июня 2003 г. № 91 |
|
|
Система стандартов безопасности труда. Безопасность электротехнических контрольно-измерительных приборов и лабораторного оборудования |
||
|
Правила устройства электроустановок, утвержденные Министерством энергетики РФ Приказ № 204 от 8 июля 2002 г.*** |
||
|
Правила обеспечения работников специальной одеждой, специальной обувью и другими средствами индивидуальной защиты, утвержденные Постановлением Министерства труда и социального развития РФ от 18 декабря 1998 г. № 51*** |
||
|
[12] Строительные нормы и правила |
Административные и бытовые здания |
|
|
[13] Технические условия ТУ 6-09-5360-88 |
Фенолфталеин |
|
|
[14] Технические условия ТУ 6-09-1181-89 |
Бумага индикаторная универсальная для определения рН 1 - 10 и 7 - 14 |
|
|
[15] Технические условия ТУ 6-09-3901-75 |
Диэтилдитиокарбамат свинца (II) |
|
|
[16] Технические условия ТУ 6-09-4711-81 |
Кальций хлорид обезвоженный (кальций хлористый) |
|
|
[17] Технические условия ТУ 6-09-3880-75 |
Магний хлорнокислый (ангидрон) |
|
|
[18] Технические условия ТУ 48-19-30-91 |
Штабики вольфрамовые сварные ос. ч. |
|
|
[19] Технические условия ТУ 6-09-05808009-262-92* |
Железо карбонильное ос. ч. 13 - 2, ос. ч. 6 - 2 |
|
|
[20] Технические условия ТУ 6-09-4128-88 |
Аскарит |
|
|
[21] Технические условия ТУ 6-09-1678-95**** |
Фильтры обеззоленные (белая, красная, синяя ленты) |
|
|
[22] Технические условия ТУ 6-09-4676-83 |
Нитраты иттрия и редкоземельных элементов (лантана, празеодима, неодима, самария, европия, гадолиния, тербия, диспрозия, гольмия, эрбия) |
|
|
[23] Технические условия ТУ 6-09-2878-84 |
Реактивы. Кислота хлорная химически чистая для анализа |
|
|
[24] Технические условия ТУ 6-09-5359-88 |
Аммоний железо (III) сульфат (1:1:2) 12-водный (квасцы железоаммонийные) чистый для анализа, чистый |
|
|
[25] Технические условия ТУ 113-38-127-92 |
Калий цианистый технический |
|
|
[26] Технические условия ТУ 6-09-5393-88 |
Олово (II) хлорид 2-водное (олово двухлористое) |
|
|
[27] Технические условия ТУ 48-6-99-87 |
Теллур особой чистоты |
|
|
[28] Технические условия ТУ 25-1819.0021-90 |
Секундомер механический |
|
|
[29] Технические условия ТУ 113-12-112-89 |
Мышьяк металлический для полупроводниковых соединений ос.ч. |
|
* На территории Российской Федерации действует ГОСТ Р ИСО 5725-1-2002.
** На территории Российской Федерации действует ГОСТ Р ИСО 5725-6-2002.
*** Документ действует на территории Российской Федерации.
**** Документ действует на территории Российской Федерации.
Ключевые слова: медь, примеси, методы анализа, общие требования, раствор, требования безопасности, стандартный образец, атомно-абсорбционный метод, массовая концентрация, раствор с известной концентрацией, градуировочный график