Методические указания
по лабораторному контролю качества продукции
общественного питания
"Порядок отбора проб и физико-химические методы испытаний"
(одобрены Министерством здравоохранения СССР 23 октября 1991 г. № 122-5/72)
(рекомендованы Министерством торговли СССР 11 ноября 1991 г. № 1-40/3805)
Раздел I, часть I
Настоящие Методические указания по лабораторному контролю качества продукции общественного питания - "Порядок отбора проб и физико-химические методы испытаний" предназначены для руководства в работе территориально-отраслевых санитарно-технологических и производственных санитарно-технологических пищевых лабораторий системы общественного питания.
Методические указания разработаны на основе нормативно-технической документации, действующей в отрасли и в смежных отраслях пищевой промышленности, унифицированных методов анализа, предусмотренных в Общесоюзных санитарно-гигиенических и санитарно-противоэпидемических правилах и нормах, методических указаниях и рекомендациях Минздрава, медико-биологических требований и санитарных норм качества продовольственного сырья и пищевых продуктов.
Методические указания включают в себя порядок отбора проб, подготовку к анализу, физико-химические методы испытаний полуфабрикатов, блюд и кулинарных изделий, методы контроля качества за правильностью ведения технологического процесса, соблюдением рецептур и требований нормативно-технической и технологической документации при приготовлении блюд и изделий, приготовление реактивов.
Методические указания состоят из двух частей, часть первая.
Раздел 1. Отбор проб
1.1. Основные положения по отбору проб продукции общественного питания
Качество полуфабрикатов, блюд и кулинарных изделий, а также сырья оценивают по результатам анализа части продукции, отобранной из партии.
Партией считается любое количество кулинарной продукции одного наименования, изготовленной предприятием за смену.
Отбор проб сырья, полуфабрикатов и кулинарных изделий, на которые разработана нормативно-техническая документация (ГОСТ, РСТ, ТУ), производят, вскрывая определенное количество транспортных единиц упаковки, оговоренное в нормативно-технической документации, и изымая часть продукции. Пробу, отобранную из отдельной единицы упаковки, называют разовой. Количество продукции в разовых пробах из каждой единицы упаковки должно быть одинаковым.
Разовые пробы соединяют, перемешивают и составляют среднюю, или общую, пробу. Средняя проба должна быть отобрана таким образом, чтобы состав ее соответствовал всей партии.
При отсутствии нормативно-технической документации на сырье и полуфабрикаты для отбора средней пробы из небольшой партии продукции вскрывают все единицы упаковки, если их не более пяти, и в более крупной - каждую вторую или третью, но не менее пяти.
Из средней пробы выделяют части для определения массы, органолептической оценки и лабораторного анализа.
Пробы сырья, полуфабрикатов, блюд и кулинарных изделий, отобранные для анализа, упаковывают в сухую, чистую тару: стеклянные банки с плотно закрывающимися крышками, металлические судки, подпергамент, целлофан, полимерную пленку. Каждая проба должна иметь этикетку с названием продукта или изделия, указанием даты и часа отбора пробы, а также номера нормативно-технической документации или рецептуры.
При выемке проб составляется акт в двух экземплярах, один из которых остается на предприятии, а другой - в лаборатории. Формы актов приведены в Рекомендациях по организации лабораторного контроля качества продукции общественного питания (Приказ Минторга СССР от 29.12.90 г. № 0184-36).
Пробы должны быть доставлены в лабораторию по возможности немедленно, но не позднее 6 ч с момента их отбора; коктейли с молочными продуктами - не позднее 2 ч, а коктейли алкогольные - не позднее 4 ч с момента их приготовления.
Для доставки проб блюд (изделий) в лабораторию лучше использовать комплект из восьми цилиндрических судков. При использовании стеклянных и полиэтиленовых банок с крышками их накрывают бумагой поверх крышек, обвязывают и пломбируют. Пломбированные судки или банки нумеруют в порядке, соответствующем записи в акте отбора проб. Мучные кондитерские и булочные изделия завертывают в пергаментную бумагу, укладывают в полиэтиленовый пакет (каждый вид изделия отдельно), обвязывают и пломбируют.
Доставленные пробы должны исследоваться, по возможности, в тот же день. Остатки проб сохраняются в холодильнике при температуре 4 - 8 °C до окончания испытаний и выдачи результатов анализа, после чего с разрешения заведующего лабораторией уничтожаются.
Органолептическую оценку в лаборатории проводят в том случае, если ее не провели на производстве или если лицо, проводящее анализ, не согласно с органолептической оценкой, приведенной в акте.
Для физико-химических исследований часть пробы превращают в однородную массу, применяя разные способы: хрупкие, крошливые полуфабрикаты, кулинарные изделия растирают в ступке или измельчают на лабораторной мельнице (кофемолке); пастообразные и легко разминающиеся полуфабрикаты, кулинарные изделия растирают в ступке, а при более плотной консистенции пропускают через мясорубку; полуфабрикаты и кулинарные изделия из мяса, рыбы и птицы пропускают через мясорубку дважды; сырые овощи измельчают на терке.
Пробы полуфабрикатов и кулинарных изделий плотной консистенции, многокомпонентные по составу, целесообразно гомогенизировать в размельчителе тканей типа РТ-1. При его отсутствии пользуются смесителем от универсальной кухонной машины (УКМ). Размельчитель предназначен для измельчения пищевых продуктов животного и растительного происхождения в жидкой среде, поэтому при измельчении некоторых блюд и полуфабрикатов добавляют определенное количество воды в зависимости от консистенции и химического состава продуктов, предусмотренных рецептурой. Измельчение производят в закрытом крышкой сосуде сначала при 4000 об/мин в течение 0,5 - 1 мин, затем при 8000 об/мин. Прибор следует включать не более чем на 5 мин. Повторное включение производят после 8 - 10-минутного перерыва.
Пробы, подготовленные к анализу, переносят в банку с притертой пробкой и берут из нее навески для испытания. Перед взятием навесок содержимое банок тщательно перемешивают. Пробы влажных продуктов, полуфабрикатов, кулинарных и кондитерских изделий хранят в холодильнике при температуре 4 - 8 °C не более суток. Перед взятием навесок пробы подогревают на водяной бане с температурой 50 - 60 °C или на воздухе до температуры 20 °C.
1.2. Определение средней массы и выхода отдельных частей полуфабрикатов, блюд и кулинарных изделий
При проверке предприятия работниками территориально-отраслевых санитарно-технологических и производственных санитарно-технологических пищевых лабораторий и других контролирующих организаций проводится определение средней массы полуфабрикатов, блюд и кулинарных изделий.
Количество полуфабрикатов, отбираемых для контроля средней массы, и допустимые отклонения в массе приведены в прил. 1.
Количество блюд и кулинарных изделий, отбираемых для определения средней массы, приведено в прил. 2.
Штучные и порционные кулинарные и мучные кондитерские изделия отбирают из разных противней или лотков и взвешивают по 10 штук на настольных циферблатных весах со шкалой до 1 кг. Преднамеренный выбор изделий не допускается. При получении заниженных результатов взвешивают еще 10 изделий. Затем производят поштучное взвешивание не менее 10 изделий на настольных циферблатных весах со шкалой до 200 г. Результаты повторных испытаний являются окончательными.
Средняя масса блюд, отобранных на раздаче, определяется путем раздельного взвешивания трех порций с последующим суммированием и делением на 3. Отклонение средней массы блюд и кулинарных изделий от установленной нормы выхода по рецептуре не допускается. Масса одного блюда (изделия) может отклоняться от нормы не более чем на ±3 %.
С целью установления правильности отпуска к блюдам растительного и сливочного масла, сметаны, сахара, порционируемых с помощью мерников или ложек, проверяют массу указанных продуктов в объеме этого инвентаря одновременным взвешиванием 10 - 20 порций.
Объем или массу отпускаемых горячих и холодных напитков (кофе, какао, чая, соков, прохладительных напитков без наполнителя и т.д.) определяют при установленной для них температуре отпуска.
Для определения количества панировки и выхода мяса, рыбы, птицы в полуфабрикатах и кулинарных изделиях с двойной панировкой (мука, льезон, сухари)*(1) взвешивают 3 - 5 изделий, затем освобождают с помощью скальпеля от панировки, снова взвешивают и рассчитывают среднюю массу. Прибавляя к средней массе массу потерь при тепловой обработке, рассчитывают фактическую массу нетто мяса, рыбы, птицы. Эту массу сравнивают с массой нетто сырья по рецептуре.
_____________
*(1) В фаршированных изделиях из мяса, рыбы, птицы и кролика количество панировки определяют в полуфабрикате.
Аналогично определяют количество мучной панировки в изделии "Печень жареная" и ее выход.
Для определения количества панировки в жареной печени, реализуемой по массе, взвешивают 3 - 5 изделий, счищают панировку, печень взвешивают и рассчитывают массу панировки в процентах к массе изделия. Полученные данные сравнивают с данными контрольных проработок, проведенных не менее трех раз.
Количество панировки и выход мяса, рыбы, птицы, кролика для изделий, в том числе фирменных, для которых нормы потерь при тепловой обработке не установлены, определяют работники лабораторий при контрольных проработках. Запись результатов взвешивания производится в акте отбора проб.
Допустимые отклонения от выхода по рецептуре приведены в табл. 1.
Допустимые отклонения от выхода по рецептуре составных частей полуфабрикатов, блюд и изделий
|
№ п/п |
Наименование полуфабрикатов, блюд, изделий |
Допустимые отклонения, % |
|
1 |
2 |
3 |
|
1. |
Салаты мясные (содержание мяса) |
±10 |
|
2. |
Студни (плотная часть) |
±10 |
|
3. |
Мясо, птица, язык, рыба заливные (масса мяса, рыбы) |
±5 |
|
4. |
Рыба, птица под майонезом |
±5 |
|
5. |
Рыба под маринадом |
±5 |
|
6. |
Супы (масса мяса, рыбы) |
±10 |
|
7. |
Горячие супы (основные овощи - картофель, капуста, свекла и т.п.) |
±10 - 15, при условии сохранения общей массы закладываемых овощей |
|
8. |
Вегетарианские супы-пюре (молоко, яйца для заправки) |
+50 |
|
9. |
Голубцы, кабачки и другие овощи, фаршированные мясом - полуфабрикат (содержание фарша) |
±5 |
|
10. |
Блинчики с разными фаршами - полуфабрикат (содержание фарша) |
±5 |
|
11. |
Блинчики с разными фаршами (готовые изделия), кроме блинчиков с творогом |
±10 |
|
12. |
Пельмени-полуфабрикат (содержание фарша) |
±5 |
|
13. |
Вареники-полуфабрикат (содержание фарша) |
±5 |
|
14. |
Компоты, коктейли и сладкие супы (плотная часть) |
±10 |
|
15. |
Мелкокусковые полуфабрикаты из мяса (отдельные кусочки) |
±15 - 25 |
Раздел 2. Физико-химические методы, применяемые при контроле полуфабрикатов, блюд и кулинарных изделий
В разделе изложены методы контроля полуфабрикатов, блюд и кулинарных изделий по физико-химическим показателям, определения химического состава продукции общественного питания и правильности проведения технологического процесса.
Продукция, на которую разработана нормативно-техническая документация, исследуется методами, изложенными в документации. Полуфабрикаты и кулинарные изделия, выпускаемые по Сборникам рецептур и другой документации, контролируются по аналогии со стандартизованной продукцией, а также методами, приведенными в данном разделе.
2.1. Определение сухих веществ или влажности
Содержание сухих веществ и влаги в полуфабрикатах, блюдах и кулинарных изделиях определяют весовыми методами (в сушильном шкафу и на приборе ВЧ) и рефрактометрическим методом.
Весовые методы основаны на выделении гигроскопической влаги из исследуемого объекта при определенной температуре. Высушивание производят до постоянной массы (арбитражный метод) или ускоренным методом при повышенной температуре в течение заданного времени.
2.1.1. Арбитражный метод (высушивание в сушильном шкафу до постоянной массы)
Аппаратура, материалы, реактивы. Шкаф сушильный лабораторный типа СЭШ или любого другого типа с автоматическим терморегулирующим устройством или лабораторным автотрансформатором (ЛАТР-1)*(2); весы лабораторные*(3); термометры лабораторные со шкалой до 150 °C и ценой деления 1 °C; эксикаторы; баня водяная или песочная; бюксы стеклянные диаметром 40 - 50 мм и высотой 25 - 45 мм или алюминиевые или чашки фарфоровые выпарительные диаметром 60 - 80 мм; палочки стеклянные оплавленные (длина палочек несколько больше диаметра бюксы, но чтобы она не мешала закрывать бюксу крышкой); сита с отверстиями 4 - 5 мм и 1 - 1,5 мм; кальций хлористый плавленый (прокаленный) или кислота серная плотностью 1,84 г/см3; кислота соляная плотностью 1,19 г/см3; вазелин технический; песок очищенный, прокаленный.
Подготовка к испытанию. Сушильные шкафы на равномерность нагрева проверяют при помощи максимальных термометров не реже одного раза в месяц.
Максимальные термометры в количестве 4 - 5 шт. размещают на полке в местах, где ставят бюксы с навесками (в центральной части полки шкафа). Расхождения между показаниями отдельных максимальных термометров допускаются не более ±2 °C.
Если температуры в различных местах шкафа отличаются более чем на ±2 °C, шкафом пользоваться нельзя. При отсутствии максимальных термометров можно проверить равномерность нагрева сушильного шкафа высушиванием в определенных намеченных участках полки 4 - 6 параллельных навесок. Расхождения между параллельными определениями при проверке работы шкафа допускаются не более 0,3 %.
При высушивании навесок конец контрольного термометра, которым измеряется температура воздуха в сушильном шкафу, должен находиться на уровне бюкс с навесками.
Показания контрольного термометра должны соответствовать фактической температуре высушивания.
При сушке вентиляционные отверстия (внизу и наверху шкафа) должны быть открыты. Бюксы ставят в определенные проверенные места полки шкафа.
Эксикаторы. Нижняя часть эксикатора должна быть заполнена прокаленным хлористым кальцием или концентрированной серной кислотой.
Пришлифованные края эксикатора смазывают техническим вазелином.
Очистка песка. Песок просеивают через сито с диаметром отверстий 4 - 5 мм, отмучивают водопроводной водой, кипятят несколько раз с соляной кислотой, разбавленной (1:1). Затем песок тщательно промывают вначале водопроводной водой до исчезновения кислой реакции (проба на лакмус), затем дистиллированной водой и высушивают. Высушенный песок просеивают через сито с диаметром отверстий 1 - 1,5 мм и прокаливают при температуре от 500 до 600 °C в течение 5 ч для удаления органических веществ. Очищенный песок хранят в чистой, плотно закрытой банке.
Проведение испытания. Высушивание образцов, спекающихся в плотную массу, производят с прокаленным песком, масса которого должна быть в 2 - 4 раза больше массы навески. Песок придает навеске пористость, увеличивает поверхность испарения, препятствует образованию на поверхности корочки, затрудняющей удаление влаги.
_____________
*(2) Пользоваться сушильными шкафами без приборов или приспособлений для поддержания температур на заданном уровне не разрешается.
*(3) Здесь и далее используются весы лабораторные общего назначения 2-го - 4-го классов точности с наибольшим пределом взвешивания 200, 500 г, 1 кг или другие с аналогичными метрологическими характеристиками.
Определение сухих веществ или влажности
|
Полуфабрикаты, изделия, блюда |
Посуда или пакеты |
Масса навески, г |
Точность взвешивания, г |
Аппарат для высушивания |
Режим высушивания |
Документ, по которому проводится определение |
|
|
температура, °С |
продолжительность, мин |
||||||
|
Рубленые полуфабрикаты из мяса, птицы, рыбы |
Фарфоровые чашки диаметром 60 - 80 мм |
5 |
0,01 |
Сушильный шкаф |
130 ± 2 |
80 |
|
|
Бульоны: костные концентрированные |
Бюксы стеклянные (металлические) или фарфоровые чашки с песком (12 - 15 г) и палочкой |
10 |
0,001 |
То же |
98 - 100 |
До постоянной массы |
ГОСТ 8756.2-82 |
|
Бульоны с желатином (мясной и куриный) |
|||||||
|
Бульон куриный костный |
|||||||
|
То же |
То же |
10 |
0,01 |
Выпаривание до видимой сухости на водяной бане, сушильный шкаф |
130 ± 2 |
30 |
МУ |
|
Соусы концентрированные |
Фарфоровые чашки диаметром 60 - 80 мм |
5 |
0,01 |
Сушильный шкаф |
130 ± 2 |
80 |
|
|
Овощные полуфабрикаты |
То же |
5 - 6 |
0,001 |
То же |
98 - 100 |
До постоянной массы |
ГОСТ 8756.2-82 |
|
Фарш голубцов |
|||||||
|
То же |
Бумажные пакеты |
5 - 6 |
0,01 |
ВЧ |
150 - 152 |
5 |
|
|
Биточки (котлеты) крупяные |
Бюксы стеклянные (металлические) диаметром 45 - 60 мм, высотой 40 - 50 мм |
5 |
0,001 |
Сушильный шкаф |
100 - 105 |
До постоянной массы |
|
|
Тесто охлажденное |
Алюминиевые бюксы диаметром 48 мм, высотой 20 мм |
5 |
0,01 |
Сушильный шкаф |
130 ± 2 |
40 |
|
|
Полуфабрикаты |
|||||||
|
То же |
Бумажные пакеты |
4 - 5 |
0,01 |
ВЧ |
155 - 160 |
5 |
ТУ 28-50-90 |
|
Полуфабрикаты тортов и пирожных |
Бюксы стеклянные (металлические) высотой 30 мм |
3 |
0,01 |
Сушильный шкаф |
130 ± 2 |
40 |
|
|
Кремы |
Стеклянные бюксы с песком и палочкой |
3 |
0,001 |
То же |
130 ± 2 |
50 |
|
|
Блинчиковая оболочка и фарш |
Бюксы стеклянные (металлические) с песком и палочкой |
5 |
0,001 |
" |
98 - 100 |
До постоянной массы |
ГОСТ 8756.2-82 |
|
Первые блюда (без выпаривания), соусы, кисели, желе, муссы, самбуки |
Бюксы стеклянные (металлические) или фарфоровые чашки с песком и палочкой |
10 |
0,001 |
" |
102 ± 2 |
То же |
МУ |
|
Первые блюда после выпаривания |
Бюксы стеклянные (металлические) или фарфоровые чашки с песком и палочкой |
5 |
0,01 |
Подсушивание на водяной бане до видимой сухости. Сушильный шкаф |
130 ± 2 |
30, а затем еще 15 |
МУ |
|
Первые блюда, соусы после выпаривания |
Бумажные пакеты |
5 |
0,01 |
ВЧ |
152 - 154 |
10 |
МУ |
|
Вторые блюда, гарниры, холодные и сладкие |
Бюксы стеклянные или фарфоровые чашки с песком и палочкой |
5 - 6 |
0,001 |
Сушильный шкаф |
102 ± 2 |
До постоянной массы |
МУ |
|
Вторые и холодные блюда |
Бумажные пакеты |
5 |
0,01 |
То же |
130 ± 2 |
90, а затем еще 15 |
МУ |
|
Вторые блюда из овощей, круп, мяса и рыбы |
То же |
5 |
0,01 |
ВЧ |
152 - 154 |
7 |
МУ |
|
Вторые блюда из бобовых и макаронных изделий |
" |
5 |
0,01 |
То же |
152 - 154 |
9 |
МУ |
|
Сдобные булочные изделия |
Алюминиевые бюксы |
5 |
0,01 |
Сушильный шкаф |
130 ± 2 |
40 |
|
|
Пирожки печеные и жареные |
Бюксы стеклянные (металлические) или фарфоровые чашки с песком и палочкой |
5 |
0,001 |
То же |
103 ± 2 |
До постоянной массы |
|
|
Основа |
|||||||
|
Фарш (начинка): мясной с луком или с рисом, из субпродуктов |
Фарфоровые чашки диаметром 60 - 80 мм |
5 |
0,01 |
То же |
130 ± 2 |
80 |
|
|
творожный |
Стеклянные бюксы с песком и палочкой |
3 - 5 |
0,001 |
" |
102 ± 2 |
До постоянной массы |
|
|
другие фарши |
Бюксы стеклянные (металлические) с песком и палочкой |
5 |
0,001 |
" |
98 - 100 |
То же |
ГОСТ 8756.2-82 |
|
Кексы, рулеты |
Бюксы стеклянные (металлические) высотой 30 мм |
3 |
0,01 |
Сушильный шкаф |
130 ± 2 |
40 |
|
|
Печенье |
То же |
3 |
0,01 |
То же |
130 ± 2 |
30 |
|
Если после перемешивания с песком продукт превращается в комок, то к навеске прибавляют 0,5 - 1 см3 дистиллированной воды и хорошо перемешивают стеклянной палочкой при подогревании на водяной бане.
Очень влажные образцы рекомендуется подсушивать на водяной или песочной бане, периодически помешивая стеклянной палочкой.
Высушивание производят в фарфоровых чашках, алюминиевых или стеклянных бюксах. Чашки или бюксы с песком и стеклянной палочкой высушивают в течение 30 мин при температуре, указанной в табл. 2, охлаждают в эксикаторе (металлические бюксы - 15 - 20 мин, стеклянные бюксы - 25 - 30 мин) и взвешивают с точностью, указанной в табл. 2.
В бюксу или чашку помещают навеску (масса указана в табл. 2) исследуемого объекта, закрывают бюксу крышкой и взвешивают на весах с указанной точностью. Затем, открыв крышку бюксы, тщательно и осторожно перемешивают навеску с песком стеклянной палочкой, равномерно распределяя содержимое по дну бюксы или чашки.
Чашку или открытую бюксу с навеской и крышку помещают в сушильный шкаф и сушат при режиме, приведенном в табл. 2. При внесении чашек или бюкс в шкаф температура в нем несколько понижается, поэтому отсчет времени производят с момента, когда термометр покажет 102 ± 2 °С. Первое взвешивание производят через 1 ч (при подсушивании на бане) или через 2 ч (без подсушивания на бане), последующие - через каждые 30 мин.
После каждого высушивания чашки или бюксы охлаждают в эксикаторе в течение 20 - 30 мин. Если уменьшение массы после первого и второго высушивания не превышает 0,002 г, высушивание заканчивают. Если при взвешивании после высушивания масса увеличится по сравнению с предыдущим значением, то для расчета принимают результат предыдущего взвешивания.
Обработка результатов. Массовую долю сухих веществ (Х, %) вычисляют по формуле
где:
m - масса бюксы со стеклянной палочкой и песком, г;
m1 - масса бюксы со стеклянной палочкой, песком и навеской до высушивания, г;
m2 - масса бюксы со стеклянной палочкой, песком и навеской после высушивания, г.
Если содержание сухих веществ в исследуемом объекте выражают в граммах, то в формулу вместо числа 100 ставят массу полуфабриката, изделия или блюда (Р).
Расхождение между результатами параллельных определений не должно превышать 0,5 %. За конечный результат принимают среднее арифметическое двух параллельных определений, вычисленное с точностью до 0,1 %. Здесь и далее результат анализа сравнивают с расчетными данными по рецептуре (теоретическими) или минимально допустимыми.
2.1.2. Ускоренный метод (высушивание в сушильном шкафу при температуре 130 °C)
Аппаратура, материалы, реактивы. Те же, что и для арбитражного метода (см. выше).
Проведение испытания. Из подготовленной пробы в фарфоровые чашки или бюксы с песком и стеклянной палочкой или без песка, предварительно высушенные до постоянной массы, взвешивают навески (масса указана в табл. 2). Для ускорения высушивания навеску распределяют ровным слоем по внутренним стенкам чашки или по дну бюксы. Вначале навески подсушивают на водяной или песочной бане до видимой сухости, периодически помешивая стеклянной палочкой. Затем чашки или открытые бюксы помещают в шкаф и досушивают навески при температуре 130 ± 2 °C в течение 30 мин. Отсчет времени высушивания производят с момента, когда термометр покажет 130 °C. После этого чашки или бюксы охлаждают в эксикаторе в течение 20 - 30 мин и взвешивают.
Если навеску высушивают при 130 °C без предварительного подсушивания на бане, то время высушивания зависит от особенностей продукта, характера и степени его измельчения. Оно строго регламентируется и устанавливается опытным путем для каждой группы изделий и проверяется арбитражным методом. Для проверки полноты высушивания навески вновь ставят в сушильный шкаф на 15 мин, после чего охлаждают и взвешивают.
Содержание сухих веществ вычисляют, как указано в арбитражном методе.
2.1.3. Ускоренный весовой метод (высушивание на приборе ВЧ)
Обезвоживание исследуемого продукта производится за счет инфракрасного излучения в аппарате, состоящем из двух соединенных между собой массивных плит круглой или прямоугольной формы.
В рабочем состоянии между плитами устанавливают зазор 2 - 3 мм. Температура греющих поверхностей контролируется двумя ртутными термометрами. Для поддержания постоянной температуры прибор снабжен контактным термометром, включенным последовательно с реле. На контактном термометре устанавливается заданная температура. Прибор включают в сеть за 20 - 25 мин до начала высушивания и нагревают до требуемой температуры.
Аппаратура, материалы. Аппарат ВЧ; весы лабораторные; часы песочные; эксикатор; шпатель; бумага ротаторная; бумага фильтровальная; фольга алюминиевая или оловянная марки ФО.
Проведение испытания. Из листов ротаторной бумаги размером 20×14 см готовят прямоугольные пакеты, а из листов размером 15×15 см - пакеты треугольной формы. При изготовлении пакета прямоугольной формы лист складывают пополам по длинной стороне, а затем открытые с трех сторон края пакета загибают на 1,5 см. Размеры готового пакета 8×11 см. Треугольные пакеты получают, складывая лист бумаги пополам по диагонали и загнув открытую часть на 1 см. В бумажные пакеты помещают 2 - 4-слойные вкладыши из фильтровальной бумаги (в зависимости от влажности изделий).
При высушивании полуфабрикатов и изделий с повышенным содержанием жира для изготовления пакетов, кроме ротаторной бумаги, используют алюминиевую фольгу. Для прямоугольных пакетов из фольги вырезают листы размером 17×11 см, складывают их по длинной стороне пополам, вкладывают пакет из ротаторной бумаги и завертывают только с длинной стороны. Для треугольных пакетов фольгу вырезают размером 14,5×14,5 см и завертывают пакет только с одной стороны.
Заготовленные пакеты высушивают в течение 3 мин при определенной температуре (табл. 2), охлаждают в эксикаторе 2 - 3 мин и быстро взвешивают.
Навеску продукта помещают в пакет, между слоями фильтровальной бумаги, быстро размазывают тонким слоем с помощью шпателя, складывают пакет, взвешивают, а затем помещают в прибор ВЧ между плитами.
При высушивании полуфабрикатов и готовых изделий с повышенной влажностью (супы, соусы) пакеты помещают на нижнюю плиту прибора, а верхнюю плиту оставляют открытой или приподнятой на 1,5 см в течение 2 - 3 мин, а затем опускают верхнюю плиту и продолжают высушивание.
Рекомендуемые режимы высушивания навески приведены в табл. 2. После высушивания пакеты помещают в эксикатор на 2 - 3 мин для охлаждения и быстро взвешивают.
Обработка результатов. Влажность (Х, %) в полуфабрикатах и изделиях рассчитывают по формуле
|
|
(2) |
где:
m - масса пакета, г;
m1 - масса пакета с навеской до высушивания, г;
m2 - масса пакета с высушенной навеской, г.
Содержание сухих веществ (X1) в полуфабрикатах (изделиях) выражают в процентах или граммах. В первом случае расчет ведут по формуле (1).
Если содержание сухих веществ выражают в граммах, то в формуле (1) вместо числа 100 ставят массу блюда или изделия, при этом учитывают разбавление водой при гомогенизации или уменьшение массы при упаривании. Например, при исследовании порции винегрета массой 150 г для гомогенизации добавили 75 см3 дистиллированной воды. Общая масса блюда после гомогенизации 225 г. При расчете количества сухих веществ (г) вместо числа 100 ставят 225.
Если перед гомогенизацией суп упарили до 300 г, то в формулу вместо числа 100 ставят число 300, так как навески для определения сухих веществ брали из упаренной пробы.
2.1.4. Рефрактометрический метод (экспресс-метод)
Данный метод применяют для производственного контроля при определении содержания сухих веществ в объектах, богатых сахарозой: сладких блюдах, напитках, соках, сиропах для промочки выпечных кондитерских изделий, сиропах для приготовления кремов, желе для отделки кондитерских полуфабрикатов.
Метод основан на зависимости между коэффициентом преломления исследуемого объекта или водной вытяжки из него и концентрацией сахарозы. Коэффициент преломления зависит от температуры, поэтому замер проводят после термостатирования призм и исследуемого раствора.
Аппаратура, материалы, реактивы. Рефрактометр лабораторный РПЛ-3, или УРЛ-У-4,2, модель 1, или РЛУ, или ИРФ-457; весы лабораторные; термостат ТС-13; баня водяная; термометр со шкалой до 100 °C с ценой деления 1 °C; пипетки вместимостью 2, 10 см3 с делениями; чашки фарфоровые выпарительные диаметром 4 - 6 см; бюксы стеклянные; палочки стеклянные оплавленные; трубочка стеклянная длиной 18 - 20 см и диаметром 0,5 - 0,6 см; колба коническая вместимостью 50 - 100 см3; стакан химический вместимостью 100 - 150 см3; воронка стеклянная диаметром 3 - 4 см; марля; бумага фильтровальная; вода дистиллированная.
Проведение испытания. Перед началом работы на штуцеры рефрактометра надевают резиновые шланги и соединяют их с термостатом, отрегулированным на 20 °C. Через 10 мин проверяют показания прибора по дистиллированной воде. На нижнюю призму рефрактометра оплавленной стеклянной палочкой наносят 1 - 2 капли дистиллированной воды, опускают верхнюю призму и через 2 - 3 мин проводят замер. Граница светотени должна быть четкой и проходить через точку пересечения нитей (перекрестие) или пунктирную линию (рефрактометр РПЛ-3). Если этого не наблюдается, то специальным торцевым ключом, прилагаемым к прибору, добиваются совпадения границы светотени с перекрестием или пунктирной линией.
Рефрактометр установлен на показатель преломления дистиллированной воды при 20 °C 1,3329, что соответствует 0 % сухих веществ.
Призмы рефрактометра вытирают сухой марлей и оплавленной стеклянной палочкой наносят 1 - 2 капли исследуемой жидкости, профильтровальной через крупнопористую фильтровальную бумагу. Опускают верхнюю призму и через 2 - 3 мин производят замер, который повторяют 2 - 3 раза, и рассчитывают среднее арифметическое.
Если определение производят не при +20 °C, то пользуются температурными поправками, приведенными в табл. 3.
Поправки на температуру для рефрактометрического определения массовой доли сухих веществ
|
Температура, °С |
Поправка при массовой доле сухих веществ в продукте, %, свыше |
|||||||||
|
0 |
5 |
10 |
15 |
20 |
25 |
30 |
40 |
50 |
60 |
|
|
|
От показания рефрактометра следует отнять |
|||||||||
|
15 |
0,27 |
0,29 |
0,31 |
0,33 |
0,34 |
0,34 |
0,35 |
0,37 |
0,38 |
0,39 |
|
16 |
0,22 |
0,24 |
0,25 |
0,26 |
0,27 |
0,28 |
0,28 |
0,30 |
0,30 |
0,31 |
|
17 |
0,17 |
0,18 |
0,19 |
0,20 |
0,21 |
0,21 |
0,21 |
0,22 |
0,23 |
0,23 |
|
18 |
0,12 |
0,13 |
0,13 |
0,14 |
0,14 |
0,14 |
0,14 |
0,15 |
0,15 |
0,16 |
|
19 |
0,06 |
0,06 |
0,06 |
0,07 |
0,07 |
0,07 |
0,07 |
0,08 |
0,08 |
0,08 |
|
|
К показаниям рефрактометра следует прибавить |
|||||||||
|
21 |
0,06 |
0,07 |
0,07 |
0,07 |
0,07 |
0,08 |
0,08 |
0,08 |
0,08 |
0,08 |
|
22 |
0,13 |
0,13 |
0,14 |
0,14 |
0,15 |
0,15 |
0,15 |
0,15 |
0,16 |
0,16 |
|
23 |
0,19 |
0,20 |
0,21 |
0,22 |
0,22 |
0,23 |
0,23 |
0,23 |
0,24 |
0,24 |
|
24 |
0,26 |
0,27 |
0,28 |
0,29 |
0,30 |
0,30 |
0,31 |
0,31 |
0,31 |
0,32 |
|
25 |
0,33 |
0,35 |
0,36 |
0,37 |
0,38 |
0,38 |
0,39 |
0,40 |
0,40 |
0,40 |
|
26 |
0,40 |
0,42 |
0,43 |
0,44 |
0,45 |
0,46 |
0,47 |
0,48 |
0,48 |
0,48 |
|
27 |
0,48 |
0,50 |
0,52 |
0,53 |
0,54 |
0,55 |
0,55 |
0,56 |
0,56 |
0,56 |
|
28 |
0,56 |
0,57 |
0,60 |
0,61 |
0,62 |
0,63 |
0,63 |
0,64 |
0,64 |
0,64 |
|
29 |
0,64 |
0,66 |
0,68 |
0,69 |
0,71 |
0,72 |
0,73 |
0,73 |
0,73 |
0,73 |
|
30 |
0,72 |
0,74 |
0,77 |
0,78 |
0,79 |
0,80 |
0,80 |
0,81 |
0,81 |
0,81 |
По шкале рефрактометра определяют коэффициент преломления или массовой доли сухих веществ. Если шкала рефрактометра градуирована на коэффициенты преломления, то по табл. 4 находят массовую долю сухих веществ.
Таблица 4
Определение массовой доли сухих веществ по показателю преломления*
|
Показатель преломления при 20 °C |
Массовая доля сухих веществ, % |
Показатель преломления при 20 °C |
Массовая доля сухих веществ, % |
Показатель преломления при 20 °C |
Массовая доля сухих веществ, % |
Показатель преломления при 20 °C |
Массовая доля сухих веществ, % |
|
1,3330 |
0,0 |
1,3456 |
8,5 |
1,3598 |
17,5 |
1,3865 |
33,0 |
|
1,3337 |
0,5 |
1,3464 |
9,0 |
1,3606 |
18,0 |
1,3883 |
34,0 |
|
1,3344 |
1,0 |
1,3471 |
9,5 |
1,3614 |
18,5 |
1,3902 |
35,0 |
|
1,3351 |
1,5 |
1,3479 |
10,0 |
1,3622 |
19,0 |
1,3920 |
36,0 |
|
1,3359 |
2,0 |
1,3487 |
10,5 |
1,3631 |
19,5 |
1,3939 |
37,0 |
|
1,3367 |
2,5 |
1,3494 |
11,0 |
1,3639 |
20,0 |
1,3958 |
38,0 |
|
1,3374 |
3,0 |
1,3502 |
11,5 |
1,3655 |
21,0 |
1,3978 |
39,0 |
|
1,3381 |
3,5 |
1,3510 |
12,0 |
1,3672 |
22,0 |
1,3997 |
40,0 |
|
1,3388 |
4,0 |
1,3518 |
12,5 |
1,3689 |
23,0 |
1,4016 |
41,0 |
|
1,3395 |
4,5 |
1,3526 |
13,0 |
1,3706 |
24,0 |
1,4036 |
42,0 |
|
1,3403 |
5,0 |
1,3533 |
13,5 |
1,3723 |
25,0 |
1,4056 |
43,0 |
|
1,3411 |
5,5 |
1,3541 |
14,0 |
1,3740 |
26,0 |
1,4076 |
44,0 |
|
1,3418 |
6,0 |
1,3549 |
14,5 |
1,3758 |
27,0 |
1,4096 |
45,0 |
|
1,3425 |
6,5 |
1,3557 |
15,0 |
1,3775 |
28,0 |
1,4117 |
46,0 |
|
1,3433 |
7,0 |
1,3565 |
15,5 |
1,3793 |
29,0 |
1,4137 |
47,0 |
|
1,3435 |
7,1 |
1,3573 |
16,0 |
1,3811 |
30,0 |
1,4158 |
48,0 |
|
1,3441 |
7,5 |
1,3582 |
16,6 |
1,3829 |
31,0 |
1,4179 |
49,0 |
|
1,3446 |
8,0 |
1,3590 |
17,0 |
1,3847 |
32,0 |
1,4200 |
50,0 |
______________
* Данные получены на основании рефрактометрии приготовленных растворов сахарозы.
Проведение испытания. Кисели и желе плодово-ягодные и молочные, муссы на желатине, самбуки, отделочные полуфабрикаты для мучных кондитерских изделий: помада, сироп, желе. Навеску подготовленной для исследования пробы массой 5 - 10 г (в зависимости от содержания сухих веществ) берут во взвешенную бюксу. Взвешивание производят с точностью до 0,01 г. Добавляют равное количество дистиллированной воды и растворяют на водяной бане с температурой 60 - 70 °C. Содержимое бюксы охлаждают до комнатной температуры, закрывают крышкой и взвешивают. Раствор рефрактометрируют, вводя температурную поправку, и рассчитывают массовую долю сухих веществ (x, %) по формуле
|
|
(3) |
где:
a - показания рефрактометра с учетом поправки на температуру, %;
m1 - масса растворенной навески, г;
m - масса навески, г.
Массу сухих веществ (Х, г) в блюде рассчитывают по формуле
|
|
(4) |
где:
х - массовая доля сухих веществ, %;
P - масса блюда, г.
За окончательный результат принимают среднее арифметическое результатов двух параллельных определений, расхождение между которыми не должно превышать 0,2 %.
При определении сухих веществ в отделочных полуфабрикатах (сахарной помаде, желе), содержащих патоку, в получаемые результаты вводят поправки в соответствии с табл. 5. Поправки даны из расчета, что каждый процент сухих веществ патоки завышает истинное содержание сухих веществ на 0,033 %.
Таблица 5
Поправки к показаниям рефрактометра для изделий, изготовленных с добавлением патоки (ГОСТ 5900-73)
|
Кол-во массовых частей патоки на 100 массовых частей сахара |
Поправка, % |
Кол-во массовых частей патоки на 100 массовых частей сахара |
Поправка, % |
|
50 |
-0,85 |
25 |
-0,46 |
|
45 |
-0,78 |
20 |
-0,37 |
|
40 |
-0,71 |
15 |
-0,27 |
|
35 |
-0,62 |
10 |
-0,16 |
|
30 |
-0,55 |
5 |
-0,07 |
Для отделочных полуфабрикатов нормируется влажность (W), которую рассчитывают по формуле
|
W = 100 - x, |
(5) |
где х - массовая доля сухих веществ, %.
Напитки с сахаром (чай, кофе). Напитки охлаждают до комнатной температуры и рефрактометрируют. Плодово-ягодные напитки сразу после подготовки наносят на призму рефрактометра.
Массовую долю сухих веществ (Х, г) рассчитывают по формуле
|
|
(6) |
где:
х - массовая доля сухих веществ, определенная рефрактометрическим методом, %;
P - объем напитка, см3.
Допускаемые отклонения в содержании сухих веществ, с учетом потерь при производстве и порционировании напитков, составляют для кофе ±1,5 %, для какао ±2,0 %.
2.2. Определение жира
2.2.1. Арбитражный метод (определение жира по обезжиренному остатку)
Метод основан на экстракции жира из исследуемого продукта серным или петролейным эфиром в экстракционном аппарате Сокслета и последующем весовом определении количества жира по разности между навеской исследуемого вещества до экстракции и после экстракции.
Аппаратура, материалы, реактивы. Весы лабораторные; экстракционный аппарат Сокслета; водяная электрическая баня; сушильный шкаф; эксикатор; фарфоровая ступка; фарфоровая чашка или часовое стекло; фильтровальная бумага; бюксы стеклянные или металлические; кальций хлористый плавленый; эфир петролейный или эфир этиловый, не содержащие перекисей.
Проведение испытания. Экстрагирование жира проводят в экстракционном аппарате Сокслета. Тщательно измельченную пробу перемешивают и, не давая отслоиться жиру, быстро отбирают с точностью до 0,001 г навеску около 5 г в небольшую фарфоровую чашку или часовое стекло, помещают в сушильный шкаф и высушивают в течение 3 - 4 ч при температуре 100 ± 2 °С. Высушенную навеску количественно переносят на заранее высушенный прямоугольный кусок фильтровальной бумаги размером 6×7 см.
Чашку или часовое стекло протирают небольшим кусочком ваты, смоченной эфиром, и эту вату присоединяют к навеске на фильтровальной бумаге.
Затем фильтровальную бумагу с навеской завертывают в виде пакета. Для предотвращения возможных потерь пакеты завертывают в несколько больший кусок предварительно высушенной фильтровальной бумаги размером 7×8 см так, чтобы линии загиба обоих пакетов не совпадали.
Бумажный пакет с навеской и ваткой помещают в высокую бюксу или на часовое стекло, высушивают для удаления эфира в сушильном шкафу в течение 10 - 15 мин и после охлаждения в эксикаторе взвешивают с точностью до 0,001 г.
Приготовленные таким образом несколько пакетов с отметкой графитным карандашом на каждом помещают в экстрактор аппарата и подвергают экстрагированию петролейным или этиловым эфиром. Эфир должен быть предварительно очищен от перекисей, высушен хлористым кальцием или сернокислым натрием и перегнан. Количество эфира, вливаемого в экстрактор аппарата, должно быть достаточным, чтобы он по сифонной трубке переливался в колбочку. Нагревание ведут на электрической водяной бане.
При перерыве в работе пакеты в экстракторе должны оставаться погруженными в эфир.
Для определения конца экстрагирования на часовое стекло наносят каплю растворителя, стекающего из экстрактора, и если на стекле после испарения эфира не остается жирового пятна, экстрагирование считают законченным. После полного извлечения жира пакеты вынимают из экстрактора, помещают в бюксы или на часовые стекла, на которых пакеты взвешивались до экстракции, и высушивают сначала 20 - 30 мин в вытяжном шкафу для удаления эфира, а затем 1,5 - 2,0 ч в сушильном шкафу при температуре 100 ± 2 °С и взвешивают с точностью до 0,001 г.
Обработка результатов. Содержание жира (Х) в процентах вычисляют по формуле
|
|
(7) |
где:
m - масса навески продукта, г;
m1 - масса бюксы или часового стекла и пакета с сухой навеской и с кусочком ваты до экстрагирования, г;
m2 - масса бюксы или часового стекла и пакета с сухой навеской и с кусочком ваты после экстрагирования, г.
Взвешивание производят с точностью до 0,01 г. За конечный результат испытаний принимают среднее арифметическое двух параллельных определений, рассчитанное с точностью до 0,1 %. Расхождение между двумя параллельными определениями не должно превышать 0,5 % при содержании жира до 12 %; 1 % - при содержании жира от 12,1 до 20 %; 1,9 % - при содержании жира от 20,1 до 30 %.
При расчете содержания жира в 1 г порции блюда (изделия) в числитель вместо 100 подставляют массу порции (изделия) с учетом упаривания или добавления воды.
2.2.2. Весовой метод с экстракцией жира в микроразмельчителе тканей
Метод основан на извлечении жира из навески растворителем в микроразмельчителе тканей типа РТ-2, фильтровании экстракта, с определением в нем жира (взвешиванием) после удаления растворителя, и предназначен для определения его в полуфабрикатах, блюдах и изделиях, указанных в прил. 1, 2.
Аппаратура, материалы, реактивы. Микроразмельчитель тканей РТ-2; шкаф сушильный лабораторный; весы лабораторные; бюксы алюминиевые диаметром 50 мм, высотой 25 - 35 мм; воронка стеклянная диаметром 30 - 40 мм; пипетка вместимостью 10 см3; стекло часовое; цилиндр мерный вместимостью 25 см3; колба мерная вместимостью 25 см3; баня водяная или песочная; часы песочные на 3 мин; эксикатор; спирт этиловый ректификованный технический высшего сорта; хлороформ технический, или метилхлороформ, или эфир петролейный; карбонат натрия безводный, или сульфат натрия безводный, или гидрофосфат натрия безводный; вата гигроскопическая или бумага фильтровальная.
Проведение испытания. Навеску исследуемой пробы в количестве 2 г взвешивают с точностью до 0,001 г в предварительно взвешенной пробирке микроразмельчителя, установив ее для устойчивости в резиновую подставку. К навеске добавляют мерным цилиндром 15 см3 экстрагирующей смеси, состоящей из хлороформа и этилового спирта (в соотношении 2:1), или из метилхлороформа и этилового спирта (в том же соотношении), или петролейного эфира. Для связывания воды, содержащейся в навеске исследуемого продукта, в пробирку добавляют карбонат натрия безводный, или гидрофосфат натрия, или сульфат натрия; при этом учитывают, что 1 г безводного карбоната или гидрофосфата натрия связывает приблизительно 1,7 г воды, 1 г безводного сульфата натрия связывает около 1,25 г воды. Чтобы гарантировать полное связывание воды, добавляют еще 2 - 2,5 г указанных реактивов. Пробирку помещают в контейнер микроразмельчителя и проводят экстракцию жира в течение 4 мин. Затем смесь накрывают часовым стеклом и оставляют на 5 - 7 мин для оседания взвешенных частиц. Пробирку снимают, раствор жира осторожно сливают в воронку с вложенным фильтром или гигроскопической ватой и фильтруют в сухую мерную колбу вместимостью 25 см3. Остатки навески промывают дважды небольшими порциями (3 - 4 см3) экстрагирующей смеси, фильтруя в ту же колбу. Содержимое колбы доводят до метки экстрагирующей смесью и хорошо перемешивают*(4). Затем отбирают пипеткой до 10 см3 экстракта, используя резиновую грушу, и переносят в предварительно высушенные и взвешенные металлические бюксы. Для удаления растворителей бюксы нагревают на водяной или песочной бане*(5) (под тягой) до исчезновения запаха растворителей. После этого бюксы с жиром помещают в сушильный шкаф и досушивают в течение 15 - 20 мин при температуре 102 ± 2 °C, охлаждают в эксикаторе и взвешивают.
_______________
*(4) При отсутствии микроразмельчителя тканей РТ-2 экстракцию жира из навески растворителем следует производить под тягой в колбе вместимостью 150 см3 в течение 1 ч при периодическом взбалтывании содержимого колбы.
*(5) Температура песочной бани должна быть не выше 120 °C.
Обработка результатов. Массовую долю (Х) жира в процентах рассчитывают по формуле
|
|
(8) |
где
m1 - масса бюксы с жиром, г;
m0 - масса пустой бюксы, г;
25 - общий объем экстракта, см3;
m - масса навески блюда (изделия), г;
10 - объем экстракта, отобранный для выпаривания, см3.
Расхождение между результатами параллельных определений не должно превышать 0,5 %. За окончательный результат испытания принимают среднее арифметическое значение, вычисленное с точностью до 0,1 %. При расчете содержания жира в г в порции блюда (изделия) в числитель вместо 100 подставляют массу порции (изделия) с учетом упаривания или добавления воды.
2.2.3. Рефрактометрический метод (ускоренный)
Метод основан на измельчении жира из навески изделия растворителем, определении коэффициентов преломления растворителя и раствора жира и вычислении его процентного содержания в изделии по соответствующей формуле и предназначен для определения содержания жира в мучных кулинарных, сдобных булочных, мучных кондитерских полуфабрикатах и изделиях, овощных полуфабрикатах.
Аппаратура, материалы, реактивы. Весы лабораторные; рефрактометр универсальный (УРЛ) с предельным показателем преломления до 1,7 или рефрактометр другой системы; шкаф сушильный электрический с контактным или техническим терморегулятором; термометр ртутный стеклянный лабораторный; часы песочные на 1, 2, 3 мин; эксикатор; пикнометр типа ПЖ2 с горловиной диаметром 6 мм, вместимостью 25, 50 см3; пипетки вместимостью 2 см3; стаканы стеклянные вместимостью 25, 50 см3; воронки стеклянные диаметром не более 40 мм; бумага фильтровальная лабораторная; ступка фарфоровая диаметром не более 70 мм с пестиком или чашка выпаривательная 1, 2 или 3; колба коническая с притертой пробкой вместимостью 25, 50, 100 см3; растворитель а-бромнафталин (монобромнафталин) с показателем преломления около 1,66; спирт этиловый технический; эфир этиловый (обезвоженный) или эфир петролейный; а-хлорнафталин с показателем преломления около 1,63.
Подготовка к испытанию. Проверка нулевой точки рефрактометра. Перед началом работы с рефрактометром проверяют нулевую точку прибора при помощи дистиллированной воды. Для этого 1 - 2 капли дистиллированной воды помещают между призмами, затем окуляр шкалы и окуляр зрительной трубы устанавливают так, чтобы визирные линии были четко видны.
Визирную линию окуляра шкалы устанавливают на 1,333 (показатель преломления дистиллированной воды при +20 °C) и в зрительную трубу наблюдают границу светотени по отношению к точке пересечения двух взаимно перпендикулярных визирных линий. При помощи специального ключа и винта ставят границу светотени на точку пересечения визирных линий, устанавливая прибор на нуль.
Проведение испытания. В фарфоровую ступку с прокаленным песком (1 - 2 г) отвешивают гомогенизированную пробу, величина которой равна (г) при содержании жира (в %): более 30 - 0,5; от 20 до 30 - 0,75; от 10 до 20 - 1,0; от 5 до 10 - 1,5; менее 5 - 2 - 5,0. Пробу подсушивают на песочной бане до полного испарения влаги. Дрожжевое тесто высушивают в аппарате ВЧ в течение 3 мин при 155 - 160 °C, охлаждают в эксикаторе, измельчают и берут навеску 2 г. Пробы сдобных булочных и мучных кондитерских изделий исследуют без предварительного подсушивания.
Навеску растирают пестиком 2 - 3 мин, затем калиброванной пипеткой приливают 2 см3 растворителя и вновь все растирают в течение 3 мин, а затем фильтруют содержимое через бумажный фильтр в пробирку. Фильтрат перемешивают стеклянной палочкой. 2 капли фильтрата наносят на призму рефрактометра, предварительно протерев призмы спиртом, термостатируют 2 - 3 мин и отсчитывают показатель преломления. Одновременно отмечают температуру с точностью до 0,1 °C. Определение повторяют 2 - 3 раза, беря за результат среднее арифметическое.
Во избежание испарения растворителя продолжительность фильтрации и определение показателя преломления должны быть не более 30 мин.
Коэффициент преломления приводят к 20 °C с внесением температурной поправки (табл. 6).
Поправку на температуру можно не вводить, если одновременно с исследуемой пробой (т.е. при одинаковой температуре) определять коэффициент преломления чистого растворителя. Температурные поправки на коэффициент преломления монобром- или монохлорнафталина и раствора жира в нем практически одинаковы, поэтому разность коэффициентов преломления растворителя и жира при одной и той же температуре равна разности коэффициентов преломления их, определенных при 20 °C.
Таблица 6
Поправка при рефрактометрическом определении показателя преломления жира и смеси жиров для температур от 15 до 35 °C
|
Температура, °С |
Поправка |
Температура, °С |
Поправка |
|
От найденного показателя преломления отнять |
|||
|
15,0 |
0,0017 |
17,5 |
0,0008 |
|
15,5 |
0,0015 |
18,0 |
0,0007 |
|
16,0 |
0,0014 |
18,5 |
0,0005 |
|
16,5 |
0,0012 |
19,0 |
0,0003 |
|
17,0 |
0,0010 |
19,5 |
0,0002 |
|
К найденному показателю преломления прибавить |
|||
|
20,5 |
0,0002 |
28,0 |
0,0028 |
|
21,0 |
0,0004 |
28,5 |
0,0030 |
|
21,5 |
0,0005 |
29,0 |
0,0031 |
|
22,0 |
0,0007 |
29,5 |
0,0033 |
|
22,5 |
0,0009 |
30,0 |
0,0035 |
|
23,0 |
0,0011 |
30,5 |
0,0037 |
|
23,5 |
0,0012 |
31,0 |
0,0038 |
|
24,0 |
0,0014 |
31,5 |
0,0040 |
|
24,5 |
0,0016 |
32,0 |
0,0042 |
|
25,0 |
0,0018 |
32,5 |
0,0043 |
|
25,5 |
0,0019 |
33,0 |
0,0045 |
|
26,0 |
0,0021 |
33,5 |
0,0047 |
|
26,5 |
0,0023 |
34,0 |
0,0049 |
|
27,0 |
0,0024 |
34,5 |
0,0050 |
|
27,5 |
0,0026 |
35,0 |
0,0052 |
Обработка результатов. Массовую долю жира (Х) в процентах вычисляют по формуле
|
|
(10) |
где
Vр - объем растворителя, взятый для извлечения жира, см3;
ρ20ж - плотность жира при 20 °C, кг/м3;
Пр - показатель преломления растворителя;
Прж - показатель преломления раствора жира в растворителе;
Пж - показатель преломления жира (табл. 7);
m - масса навески продукта, г.
Массовую долю жира (Х1) в процентах в пересчете на сухое вещество вычисляют по формуле
|
|
(11) |
где W - массовая доля влаги в исследуемом продукте, %.
За окончательный результат испытания принимают среднее арифметическое результатов двух параллельных определений, вычисленное с точностью до 0,01. Предел возможных значений погрешности измерений 0,5 % (Р = 0,95).
Примечания.
1. При вычислении массовой доли жира пользуются показателями преломления и плотности жиров (табл. 7).
2. Если в исследуемом продукте находится смесь жиров, то показатель преломления и плотность допускается определять расчетным путем.
Показатель преломления смеси жиров допускается также определять экстрагированием жира из исследуемого продукта следующим образом: 5 - 10 г измельченного продукта смешивают с 15 - 20 см3 этилового или петролейного эфира, хлороформа или четыреххлористого углерода, взбалтывают в течение 10 мин, вытяжку профильтровывают в колбу, растворитель полностью отгоняют, остаток подсушивают в сушильном шкафу при температуре 100 - 105 °C в течение 30 мин и определяют показатель преломления смеси жиров с учетом поправки на температуру.
3. Для неизвестных жира и смеси жиров плотность принимают равной 930 кг/м3.
4. Если исследуемый продукт содержит более 5 % воды, то ступку с навеской помещают в сушильный шкаф и подсушивают навеску при температуре 100 - 105 °C в течение 30 мин, затем в ступку, после ее охлаждения до комнатной температуры, приливают микропипеткой растворитель.
5. При хорошем растирании навески с растворителем в ступке, когда смесь перенесена на фильтр, разрешается стекающие из воронки капли раствора жира в растворителе наносить на призму рефрактометра, не дожидаясь, когда профильтруется вся смесь.
Показатели преломления и плотности жиров при 20 °C
|
Наименование жиров |
Плотность, кг/м3 |
Показатель преломления |
|
Жиры типа "Шоклин" |
930,0 |
1,4642 |
|
Масло какао |
937,0 |
1,4647 |
|
Масло кокосовое |
928,0 |
1,4567 |
|
Жир кондитерский |
928,0 |
1,4674 |
|
Концентраты фосфатидные |
922,0 |
1,4746 |
|
Масло коровье |
930,0 |
1,4637 |
|
Масло кукурузное |
920,0 |
1,4745 |
|
Жир кулинарный |
926,0 |
1,4724 |
|
Масло кунжутное |
918,0 |
1,4730 |
|
Маргарин |
928,0 |
1,4690 |
|
Масло орехов: |
|
|
|
арахиса |
914,0 |
1,4704 |
|
кешью |
912,0 |
1,4692 |
|
миндаля |
912,0 |
1,4707 |
|
фундука |
912,0 |
1,4706 |
|
Масло ядра абрикосовой косточки |
918,0 |
1,4715 |
|
Масло подсолнечное |
924,0 |
1,4736 |
|
Жир свиной топленый |
917,0 |
1,4712 |
|
Масло соевое |
922,0 |
1,4756 |
2.2.4. Метод определения жира с предварительным гидролизом крахмала
Метод основан на извлечении жира из предварительно гидролизованной навески изделия растворителем и определении количества жира взвешиванием после удаления растворителя из определенного объема полученного раствора.
Методом пользуются для определения массовой доли жира в мучных кондитерских изделия, отделочных и выпеченных полуфабрикатах (ГОСТ 5899-85).
Аппаратура, материалы, реактивы. Весы лабораторные; шкаф сушильный электрический с контактным или техническим терморегулятором; часы песочные на 1, 2, 3 мин; центрифуга лабораторная; электроплитка; эксикатор; баня водяная; колбы вместимостью 100, 250 см3; цилиндр вместимостью 100 см3; пипетки вместимостью 20, 250 см3; холодильник шариковый; холодильник с прямой трубкой; стаканы стеклянные вместимостью 25, 50 см3; воронки стеклянные; вата медицинская гигроскопическая; бумага фильтровальная лабораторная; груша резиновая; кислота соляная, раствор с массовой долей 1,5 %; кислота серная, раствор с массовой долей 5 %; хлороформ (трихлорметан) или этилен хлористый (дихлорэтан) плотностью 1252,0 - 1253,5 кг/м3; аммиак водный; фенолфталеин, спиртовой раствор с массовой долей 1 %.
Проведение испытания. Навеску измельченного исследуемого продукта в количестве 10 г (при содержании жира в изделиях свыше 10 % навеска может быть уменьшена до 5 г) взвешивают с точностью до 0,001 г, помещают в коническую колбу вместимостью 250 см3, приливают 100 см3 1,5-%-ной соляной кислоты (или 100 см3 5-%-ной серной кислоты), кипятят в колбе с обратным холодильником на слабом огне 30 мин. Затем колбу охлаждают водой до комнатной температуры, вносят 50 см3 хлороформа, плотно закрывают хорошо пригнанной пробкой, энергично взбалтывают в продолжение 15 мин, выливают содержимое в центрифужные пробирки и центрифугируют в продолжение 2 - 3 мин. В пробирке образуется три слоя; верхний (водный) слой удаляют. Пипеткой, снабженной резиновой грушей, отбирают хлороформный раствор жира и фильтруют его в сухую колбу через небольшой ватный тампон, вложенный в узкую часть воронки, причем кончик пипетки должен при этом касаться ваты. 20 см3 фильтрата помещают в предварительно доведенную до постоянной массы и взвешенную с точностью до 0,001 г колбу вместимостью примерно 100 см3.
Фильтрация и отбор должны проводиться в течение 2 мин, хлороформ из колбы отгоняют на водяной бане, пользуясь холодильником с прямой трубкой. Оставшийся в колбе жир сушат до постоянной массы обычно 1 - 1,5 ч при температуре 100 - 105 °C, охлаждают в эксикаторе 20 мин и взвешивают колбу с точностью до 0,001 г.
Допускается следующий способ расслаивания. После гидролиза в охлажденную колбу добавляют 5 см3 раствора аммиака (плотностью 910,0 кг/см3), 50 см3 хлороформа, взбалтывают в течение 15 мин и оставляют на 1 ч для отслаивания. За это время полностью отделяется и становится четко видимым нижний хлороформный слой. Если расслаивания не произойдет, добавляют еще 2 - 3 см3 аммиака, следя за тем, чтобы реакция по фенолфталеину оставалась кислой. После расслаивания отбор, фильтрацию, отгонку хлороформного слоя и высушивание жира ведут, как описано выше.
Обработка результатов. Массовую долю жира (Х) в процентах в пересчете на сухое вещество вычисляют по формуле
|
|
(12) |
где:
m1 - масса колбы с высушенным жиром, г;
m2 - масса пустой колбы, г;
50 - объем хлороформа, взятый для растворения жира, см3;
m - масса навески, г;
20 - объем хлороформного раствора жира, взятый для отгона, см3;
W - массовая доля влаги в исследуемом изделии, %.
Результаты параллельных определений вычисляют с точностью до 0,01 г, окончательный результат округляют до 0,1 г.
За окончательный результат испытания принимают среднее арифметическое результатов двух параллельных определений, допустимые расхождения между которыми в одной лаборатории не должны превышать по абсолютной величине 0,3 %, а выполненных в разных лабораториях - 0,5 %.
2.2.5. Метод Гербера
Метод основан на разрушении белков исследуемого продукта концентрированной серной кислотой и растворении жира в изоамиловом спирте.
Методом Гербера пользуются для определения жира в полуфабрикатах из мяса, творога, кулинарных изделиях, мучных кондитерских изделиях, молоке и молочных продуктах, сухих продуктах детского и диетического питания.
При определении жира используют молочные или сливочные жиромеры, отличающиеся размерами и градуировкой. Объем деления в молочных жиромерах равен 0,1 %, или 0,01133 г жира в продукте, пределы измерений - от 0 до 6 и от 0 до 7 весовых процентов. В сливочных жиромерах объем двух делений соответствует 1 % жира в продукте при навеске 5 г, и их используют, если содержание жира превышает 10 %.
Аппаратура, материалы, реактивы. Центрифуга; водяная баня для жиромеров; жиромеры молочные или сливочные с резиновыми пробками; автопипетки на 1 и 10 см3; штатив для жиромеров; термометр ртутный стеклянный лабораторный с пределами измерения от 0 до 100 °C; стаканы химические или чашки фарфоровые вместимостью 50 см3; воронки с коротким отростком; стеклянные палочки; кислота серная плотностью 1,50 - 1,65; 1,60 - 1,65; 1,80 - 1,81; 1,81 - 1,82 г/см3; спирт изоамиловый.
Проведение испытания. Кулинарные изделия. В фарфоровую чашку или стеклянный стакан берут навеску подготовленной пробы (с точностью до 0,01 г): первого блюда - 5 - 7, второго - 3 - 5, второго блюда с влажностью до 10 % - 2 - 2,5, соусов красного, белого - 5, соуса сметанного - 2, сладкого блюда - 5, холодного блюда - 5. К навеске добавляют 10 см3 серной кислоты плотностью 1,51 - 1,65 г/см3, нагревают на водной бане до полного растворения навески, после чего сливают жидкость в жиромер через воронку с коротким тубусом. Сливать следует так, чтобы горлышко жиромера оставалось сухим. Стакан ополаскивают 2 - 3 раза небольшим количеством серной кислоты, сливая ее в жиромер. Затем в жиромер вливают 1 см3 изоамилового спирта, добавляют такое количество серной кислоты, чтобы она не доходила на 5 - 10 мм до горлышка жиромера, закрывают сухой резиновой пробкой и, обернув полотенцем, осторожно встряхивают. Затем жиромер, перевернув пробкой вниз, помещают на 5 мин в водяную баню с температурой 65 ± 2 °С, периодически встряхивая. При этом продолжается растворение навески. Вынув жиромер из водяной бани, его обтирают, вставляют расширенной частью в патроны центрифуги, располагая симметрично один против другого, и центрифугируют 5 мин со скоростью 1300 - 1500 об/мин. Затем жиромер снова помещают на 5 мин в водяную баню с температурой 65 ± 2 °С и, вынув из бани, производят отсчет делений, занимаемых выделившимся жиром. Для этого жиромер держат вертикально так, чтобы верхняя граница жира находилась на уровне глаз. Двигая пробку вверх и вниз, устанавливают нижнюю границу столбика жира на целом делении шкалы жиромера и от него отсчитывают число делений до нижней точки мениска жирового столбика. Граница раздела жира и кислоты должна быть резкой, а столбик жира прозрачным. Если в градуированной части жиромера образовалось буроватое кольцо (пробка) или в столбике жира оказались примеси, анализ проводят повторно.
Если при описанном режиме извлечение жира будет неполным, центрифугирование и нагревание жиромера в водяной бане повторяют 2 - 3 раза.
Массу жира (Х, г) в порции блюда вычисляют по формулам
для молочного жиромера
для сливочного жиромера
где:
a - количество мелких делений жиромера, занятых выделившимся жиром;
m - масса исследуемого блюда (изделия), г;
m1 - масса навески, г;
5 - величина навески, на которую рассчитан сливочный жиромер, г;
2 - коэффициент пересчета делений сливочного жиромера в процентах;
100 - перевод результата из процента в граммы.
Расхождение между параллельными определениями не должно превышать 0,5 % жира. Полученные данные сравнивают с нормой жира по рецептуре с учетом коэффициента открываемости жира этим методом (табл. 8, 9).
Творожные полуфабрикаты и творожные изделия. В сливочный жиромер отвешивают 5 г продукта, добавляют 5 см3 воды. По стенке слегка наклоненного жиромера вливают 10 см3 серной кислоты плотностью 1,81 - 1,82 г/см3 (при анализе сладких творожных изделий - плотностью 1,80 - 1,81 г/см3) и 1 см3 изоамилового спирта. Закрыв жиромер пробкой, его 2 - 3 раза перевертывают. Подогревание жиромеров перед центрифугированием и дальнейший анализ проводят, как указано выше, расчет по формулам (13, 14).
Молочные коктейли. В молочный жиромер отвешивают 5 г коктейля, приливают 16 см3 серной кислоты плотностью 1,50 - 1,55 г/см3 так, чтобы уровень жидкости был на 4 - 6 см ниже основания жиромера. Затем добавляют 1 см3 изоамилового спирта, закрывают жиромер пробкой и проводят определение, как указано выше, применяя четырехкратное центрифугирование (со скоростью не менее 1000 об/мин) и подогревание на водяной бане при температуре 65 ± 2 °C по 5 мин перед каждым центрифугированием и отсчетом после последнего центрифугирования. Расчет содержания жира проводят по формуле (13). За норму жира принимают суммарное содержание его в продуктах, входящих в коктейль. Допустимые отклонения в содержании жира ±10 %.
Мучные кондитерские изделия. Навеску сдобных булочных или мучных кондитерских изделий (1 - 2 г) отвешивают с точностью до 0,001 г в молочный жиромер. Вливают в жиромер 6 см3 теплой воды (около 30 °C) и дают постоять 1 мин. Добавляют 1 см3 изоамилового спирта и столько серной кислоты плотностью 1,65 г/см3, сколько необходимо для заполнения жиромера до основания шейки. Закрыв жиромер пробкой, его 2 - 3 раза перевертывают и ставят на 10 мин в водяную баню с температурой 65 - 70 °C, после чего центрифугируют 5 мин и снова ставят на 5 мин в водяную баню.
Массовую долю жира (Х, %) на сухое вещество рассчитывают по формуле
|
|
(15) |
где:
a - количество мелких делений жиромера, занятых выделившимся жиром;
m - масса навески продукта, г;
W - влажность изделия, %.
При использовании сливочного жиромера навеску увеличивают до 5 г. Отсчет по шкале жиромера соответствует процентному содержанию жира в исследуемом продукте. При меньшей навеске количество делений следует умножить на коэффициент 5.
Фарш для пельменей. От средней пробы полуфабриката отделяют 200 г. Фарш дважды пропускают через мясорубку и перемешивают. Во взвешенную фарфоровую чашку диаметром 6 - 8 см отвешивают 1 - 3 г фарша (в зависимости от содержания жира) и добавляют 5 см3 серной кислоты плотностью 1,5 г/см3. Содержимое чашки осторожно нагревают при помешивании стеклянной палочкой в течение 5 - 10 мин, не допуская кипения. Если после этого остаются нерастворившиеся комочки, то добавляют еще 2 - 3 см3 кислоты и снова подогревают до образования однородной массы.
В молочный бутирометр наливают 5 см3 серной кислоты, осторожно переносят туда обработанную навеску, остатки навески смывают еще 5 см3 серной кислоты. В жиромер добавляют 2 - 4 см3 изоамилового спирта и закрывают его резиновой пробкой высотой с горлышко. Жиромер обертывают полотенцем и переворачивают 2 - 3 раза для перемешивания смеси, помещают на 10 мин на водяную баню, температура воды в которой 65 - 70 °C, центрифугируют при 800 - 1000 об/мин. в течение 5 мин, снова ставят на водяную баню на 5 мин и отсчитывают число делений, занимаемых столбиком жира.
Взбалтывание, нагрев и центрифугирование продолжают до тех пор, пока высота столбика жира не перестанет увеличиваться. Для расчета берут максимальное значение высоты столбика жира.
Массовую долю жира (в %) рассчитывают по формуле (13), приняв Р = 100.
Ниже приведены количества жира, открываемые методом Гербера в блюдах и кулинарных изделиях (табл. 8).
Количество жира, открываемое методом Гербера в блюдах и кулинарных изделиях
|
Наименование блюд (изделий) |
Количество жира, %, не менее |
|
Холодные блюда |
|
|
Салаты мясные и овощные |
80 |
|
Салаты рыбные |
90 |
|
Салат из зеленого лука со сметаной |
90 |
|
Салаты из свежей и квашеной капусты |
70 |
|
Салаты из свежих огурцов, помидоров, редиса со сметаной |
85 |
|
Свекла со сметаной |
85 |
|
Салат из сырой тертой моркови со сметаной |
90 |
|
Паштеты из печени |
90 |
|
Винегреты |
80 |
|
Редька тертая со сметаной |
90 |
|
Редька тертая с маслом |
85 |
|
Творог со сметаной и сахаром |
95 |
|
Супы |
|
|
Супы-пюре из круп |
80 |
|
Супы-пюре овощные |
70 |
|
Супы картофельные |
80 |
|
Супы картофельные с овощами, крупой, бобовыми, |
75 |
|
макаронными изделиями |
|
|
Супы с макаронными изделиями |
80 |
|
Суп рисовый молочный |
75 |
|
Щи, борщи, рассольники |
70 |
|
Супы молочные с манной, пшеничной, ячневой и другими крупами и макаронными изделиями |
80 |
|
Окрошка |
80 |
|
Мясные блюда |
|
|
Азу по-татарски, жаркое по-домашнему (мясо, соус и овощи) |
70 |
|
Бефстроганов (мясо и соус) |
80 |
|
Гуляш из говядины (мясо и соус) |
75 |
|
Голубцы с мясом и рисом (с соусом) |
75 |
|
Мясо, тушенное крупными и порционными кусками (мясо и соус) |
80 |
|
Антрекот, лангет из мяса I категории |
45*(1) |
|
Антрекот, лангет из мяса II категории |
55*(1) |
|
Ромштекс из мяса I категории |
55*(1) |
|
Ромштекс из мяса II категории |
65*(1) |
|
Колбаса жареная |
75*(1) |
|
Печень по-строгановски и печень жареная в сметанном соусе (печень и соус) |
70 |
|
Рагу из баранины (мясо и соус) |
70 |
|
Котлеты, биточки, шницели, тефтели из мяса I категории |
60*(1) |
|
Котлеты, биточки, шницели, тефтели из мяса II категории |
70*(1) |
|
Рыбные блюда |
|
|
Котлеты, биточки, тефтели |
70*(1) |
|
Рыба, жаренная куском (непластованная) |
55*(1) |
|
Филе, жаренное с кожей и хребтовой костью |
55*(1) |
|
Филе, жаренное с кожей без хребтовой кости |
70*(1) |
|
Осетрина, жаренная куском |
50*(1) |
|
Овощные блюда |
|
|
Котлеты и запеканки овощные |
75*(2) |
|
Овощи жареные |
75 |
|
Капуста отварная с маслом или соусом |
80 |
|
Капуста тушеная квашеная |
75 |
|
Капуста тушеная свежая |
80 |
|
Картофельное пюре |
90 |
|
Картофель жареный (основным способом) |
80 |
|
Овощи отварные, тушеные |
80 |
|
Овощи в молочном или сметанном соусе (припущенные) |
80 |
|
Картофель и овощи, тушенные в соусе |
70 |
|
Блюда из круп и бобовых |
|
|
Каши: |
|
|
пшеничная, рисовая, манная |
80 |
|
перловая, овсяная, гречневая |
70 |
|
Бобовые (чечевица, горох, фасоль) с жиром |
70 |
|
Запеканки |
80*(4) |
|
Биточки, котлеты |
70*(4) |
|
Блюда и изделия из творога |
|
|
Сырники (полуфабрикат) |
90 |
|
Сырники жареные |
75*(5) |
|
Мучные блюда и изделия и блюда из макаронных изделий |
|
|
Оладьи |
80*(5) |
|
Блины |
80*(3) |
|
Блинчики с мясом |
80*(4) |
|
Макароны отварные |
75 |
|
Вермишель отварная |
85 |
|
Соусы |
|
|
Белые |
75 |
|
Сметанные и молочные |
80 |
|
Красные, грибные |
70 |
_____________
*(1) Коэффициент учитывать при расчете рецептур и анализе содержания жира во всем блюде в случае определения количества основных пищевых веществ и энергетической ценности блюд.
*(2) Коэффициент учитывать для основного изделия. При отпуске котлет и запеканок с соусом учитывать коэффициент соуса (см. табл. 8).
*(3) Коэффициент учитывать для основного изделия при расчете рецептур и анализе содержания жира во всем блюде; при этом жир, используемый для выпечки изделий, не учитывать.
*(4) Коэффициент учитывать при расчете и анализе содержания жира во всем блюде; при этом жир, используемый для выпечки блинчиков, не учитывать.
*(5) Коэффициент учитывать для основного изделия при расчете рецептур и анализе содержания жира во всем блюде.
Учитывая, что лаборатории контролируют более расширенный ассортимент изделий, для получения сопоставимых данных следует руководствоваться табл. 9.
|
Изделия, на которые нормативы определяемости жира не установлены |
Изделия, к нормативу которого они могут быть отнесены по аналогии |
|
Холодные блюда |
|
|
Паштет мясной |
Паштет из печени |
|
Супы |
|
|
Холодные борщи |
Окрошка |
|
Мясные блюда |
|
|
Субпродукты в соусе, почки по-русски |
Азу |
|
Печень тушеная |
Гуляш |
|
Поджарка |
Бефстроганов |
|
Бифштекс рубленый |
Котлеты, биточки, шницели из мяса II категории |
|
Зразы рубленые |
|
|
Кабачки, баклажаны, перец, помидоры, фаршированные мясом |
Голубцы с мясом и рисом |
|
Плов |
Гуляш |
|
Овощные блюда |
|
|
Овощи припущенные |
Овощи тушеные |
|
Рагу из овощей |
Овощи тушеные |
|
Картофель отварной, картофель в молоке |
Картофельное пюре |
|
Каша из тыквы |
Картофельное пюре |
|
Кабачки, голубцы, фаршированные овощами, перец, фаршированный овощами |
Овощи жареные |
|
Икра баклажанная, из кабачков, зеленых помидоров |
Овощи тушеные |
|
Блюда из творога |
|
|
Запеканки, пудинги из творога |
Сырники |
2.2.6. Ускоренный экстракционно-весовой метод определения жира
Метод основан на экстракции жира смесью хлороформа и этилового спирта в фильтрующей длительной воронке с последующим определением его массы в полученном экстракте после удаления растворителя.
Методом пользуются для определения жира в фарше мясном, концентрированных бульонах, соусах (полуфабрикатах), в полуфабрикатах мясных, овощных котлетах и запеканках (ГОСТ 23042-86).
Аппаратура, материалы, реактивы. Прибор для экстракции жира; весы лабораторные; шкаф сушильный электрический; пипетки с резиновой грушей; мерный цилиндр вместимостью 100 см3; бюксы стеклянные; водоструйный насос; водяная баня; хлороформ; этанол.
Подготовка к испытанию. Готовят экстрагирующую смесь, смешивая два объема хлороформа с одним объемом этилового спирта.
Собирают установку для экстрагирования жира (рис. 1). В приемник вносят 2 - 3 см3 экстрагирующей смеси.

Рис. 1. Прибор для экстракции жира
1 - Фильтрующая делительная воронка; 2 - стеклянный впаянный фильтр ПОР-40; 3 - приемник
Примечание. После многократного использования фильтрующей делительной воронки в случае замедления скорости фильтрации проводят регенерацию стеклянного фильтра: промывают воронку водой, заливают 50 - 10 см3 смеси из равных объемов азотной и серной кислот и оставляют с открытым краном для стекания смеси. Через 10 - 12 ч воронку промывают проточной водопроводной водой, соединяют с приемником и снова промывают при включенном отсосе последовательно 30 см3 дистиллированной воды, а затем 30 см3 экстрагирующей смеси.
Проведение испытания. Подготовленную пробу дополнительно гомогенизируют при помощи размельчителя тканей. При необходимости продукты, содержащие животный жир, предварительно подогревают.
Навеску средней пробы (2 г), взвешенную с точностью до 0,001 г, помещают в делительную воронку со стеклянным фильтром, приливают по 10 см3 экстрагирующей смеси хлороформа с этанолом в соотношении 1:2. Экстракцию проводят в течение 2 мин при встряхивании. Экстракт с помощью водоструйного насоса отсасывают в приемник, а из него в мерный цилиндр вместимостью 100 см3. Остаток навески аналогичном способом экстрагируют еще два раза. Затем воронку и приемник промывают 20 см3 экстрагирующей смеси. Промывные жидкости собирают в мерный цилиндр и замеряют общий объем экстракта. Из цилиндра отбирают пипеткой с грушей по 20 см3 экстракта и переносят в предварительно высушенные и взвешенные бюксы.
Растворитель выпаривают на водяной бане до исчезновения запаха и высушивают навеску жира в сушильном шкафу при температуре 103 ± 2 °C до постоянной массы.
Массовую долю жира (Х, %) рассчитывают по формуле
где:
m1 - масса пустой бюксы, г;
m2 - масса бюксы с жиром, г;
m - масса навески, г;
V - общий объем экстракта, см3;
20 - объем экстракта для определения жира, см3.
Пробы овощных котлет или запеканок предварительно обезвоживают спиртом. Для этого навеску (3 г) переносят с помощью этилового спирта (10 см3) в делительную воронку со стеклянным фильтром, затем осторожно круговыми движениями перемешивают навеску со спиртом и оставляют на 10 мин до осветления жидкости над осадком. Экстракт из воронки сливают в приемник, заливают в воронку экстрагирующую смесь хлороформ-этанол и проводят экстракцию жира, удаление растворителей и высушивание бюксы, как описано выше.
Для отделения нелипидных примесей в высушенную бюксу дважды приливают по 10 см3 хлороформа и через 5 мин сливают хлороформный раствор.
Бюксу с нерастворенным осадком подсушивают в сушильном шкафу 5 мин при температуре 103 ± 2 °C, охлаждают в эксикаторе и взвешивают.
Массовую долю жира (Х, %) вычисляют по формуле
|
|
(17) |
где m3 - масса бюксы с нелипидными примесями. Остальные обозначения те же, что в формуле (16).
При определении жира экстракционно-весовым методом в расчетах минимально допустимого содержания его по рецептуре учитывают потери (в % общего содержания чистого жира в г, введенного в блюдо) в размерах, указанных в табл. 10.
Таблица 10
|
Наименование |
Потери жира, % |
|
Холодные блюда |
5 |
|
Первые блюда и соусы |
10 |
|
Вторые блюда: |
|
|
жареные, тушеные |
15 |
|
отварные, запеченные |
10 |
|
Гарниры |
15 |
|
Сладкие блюда, в рецептуру которых входят жиросодержащие продукты |
10 |
2.2.7. Определение вида жира по числу Рейхерта-Мейссля
Метод основан на извлечении жира из навески продукта растворителем (этиловым или петролейным эфиром), отгоне растворителя и высушивании жира. Определение числа Рейхерта-Мейссля основано на омылении жира и перегонке выделившихся летучих растворимых в воде жирных кислот с последующим титрованием их щелочью.
Число Рейхерта-Мейссля показывает, сколько сантиметров кубических 0,1 моль/дм3 (0,1 н) щелочи требуется для нейтрализации растворимых в воде летучих жирных кислот, отогнанных на 5 г жира. Число Рейхерта-Мейссля для маргарина 0,5 - 0,6, для сливочного масла 18 - 35. Опытным путем установлено, что из навески жира, равной 5 г, в 110 см3 дистиллята (при определенных условиях перегонки) переходят почти все летучие кислоты.
Метод применяют при определении вида жира в кондитерских кремах и жира, используемого для поливки вторых блюд.
Аппаратура, материалы, реактивы. Весы лабораторные; колба плоскодонная вместимостью 300 см3; чашка фарфоровая диаметром 5 - 7 см; шкаф сушильный лабораторный с терморегулятором; пипетка вместимостью 2 см3; цилиндр измерительный вместимостью 25 см3; холодильник стеклянный лабораторный; пробки резиновые; пробки корковые; колбы мерные вместимостью 100, 110 см3 и 1 дм3; каплеуловитель стеклянный лабораторный; колба коническая вместимостью 250 - 300 см3; воронка стеклянная; плитка электрическая; сетка асбестовая; гидроксид натрия 500 г/дм3 и 0,1 моль/дм3; растворы; глицерин; кислота серная (25 см3 концентрированной кислоты разводят водой до 1 дм3); пемза; эфир петролейный; спирт этиловый; эфир этиловый; фенолфталеин; спиртовой раствор с массовой долей 1 %; бумага фильтровальная; сульфат натрия безводный.
Подготовка к испытанию. Для извлечения жира берут навеску крема в количестве 12 - 15 г в фарфоровую чашку, хорошо растирают, переносят с помощью 40 - 50 см этилового или петролейного эфира в коническую колбу, добавляют 5 - 6 г безводного сульфата натрия, закрывают ее пробкой и взбалтывают 3 - 5 мин. Жидкую часть фильтруют в фарфоровую чашку. Затем отгоняют растворитель на водяной бане, жир подсушивают в сушильном шкафу при температуре 102 ± 2 °С в течение 1 ч, охлаждают и используют для определения числа Рейхерта-Мейссля. Жир, используемый для поливки вторых блюд, только досушивают в сушильном шкафу.
Проведение испытания. В плоскодонную колбу вместимостью 300 см3 отвешивают точно 5 г жира, прибавляют цилиндром 23 см3 глицерина (2 см3 остается на его стенках) и 2 см3 500 г/дм3 раствора едкого натра.
Жир омыляют при осторожном нагревании колбы на электрической плитке или на пламени газовой горелки при постоянном взбалтывании содержимого колбы.
Нагревание и перемешивание прекращают, когда смесь станет совершенно прозрачной, что указывает на окончание омыления. Раствор охлаждают до 80 - 90 °C и добавляют 90 см3 воды такой же температуры. Мыло растворяют, перемешивая содержимое колбы круговыми движениями, а при необходимости нагревают до кипения. При полном омылении должен получиться совершенно прозрачный раствор. В случае неполного омыления анализируемого жира вместо прозрачного раствора образуется эмульсия, и омыление необходимо начать снова.
К полученному раствору доливают 50 см3 разбавленной серной кислоты, вносят 0,6 - 0,7 г грубо измельченной пемзы (для устранения толчков при кипении) и приступают к отгонке летучих кислот. Для этого колбу соединяют с холодильником, подставляя для сбора дистиллята мерную колбу вместимостью 110 см3. Отгонку ведут с такой скоростью, чтобы получить 110 см3 дистиллята за 18 - 21 мин. Температура дистиллята 20 - 23 °C. Как только дистиллят дойдет до метки 110 см3, перегонку прекращают, колбу с дистиллятом охлаждают 10 мин в струе проточной воды с температурой 15 °C.
Закрыв колбу пробкой, жидкость осторожно взбалтывают, затем фильтруют через сухой фильтр в мерную колбу вместимостью 100 см3. Фильтрат (100 см3) переливают в коническую колбу вместимостью 250 см3 и титруют 0,1 моль/дм3 (0,1 н) раствором щелочи в присутствии 2 - 3 капель индикатора фенолфталеина до появления розового окрашивания, не исчезающего в течение 1 мин.
Обработка результатов. Число см3 0,1 моль/дм3 (0,1 н) раствора щелочи, пошедших на нейтрализацию 100 см3 дистиллята, умножают на 1,1, так как вместо 110 см3 фильтрата, отогнанных из 5 г жира, на титрование было взято лишь 100 см3.
По величине числа Рейхерта-Мейссля делают вывод о полной замене сливочного масла маргарином или добавлении маргарина к сливочному маслу.
Пример. На нейтрализацию 100 см3 фильтрата, полученного после отгонки летучих жирных кислот из крема, пошло 22,4 см3 0,1 моль/дм3 (0,1 н) раствора щелочи. Число летучих растворимых в воде жирных кислот равно 22,4 × 1,1 = 24,64 см3.
Заключение. Для изготовления крема использовано сливочное масло.
2.2.8. Обнаружение замены сливочного масла другими видами жиров
2.2.8.1. Определение вида жира люминесцентным методом
Методом люминесцентного анализа определяют вид жира в кондитерских кремах, изделиях, гарнирах, супах и жира, используемого для поливки вторых блюд.
Метод основан на извлечении жира из продуктов растворителем, отгоне растворителя и определении вида жира в приборе ЛПК-1.
Аппаратура, материалы, реактивы. Люминоскоп ЛПК-1; весы лабораторные; шкаф сушильный лабораторный с терморегулятором; баня водяная; чашки фарфоровые диаметром 7 - 9 см; ступка фарфоровая с пестиком диаметром 7 - 9 см; цилиндры измерительные вместимостью 50 и 100 см3; колба коническая с притертой пробкой вместимостью 250 см3; стаканы химические вместимостью 100 - 150 см3; палочка стеклянная; воронка стеклянная диаметром 4 - 5 см; бумага фильтровальная; эфир этиловый или петролейный; сульфат натрия безводный, или гидрофосфат натрия безводный, или карбонат натрия безводный.
Подготовка к испытанию. В зависимости от содержания жира берут навески в количестве: крема - 4 - 5 г, кондитерского изделия (измельченного после удаления корочек) - 30 - 50 г, гарнира 30 - 40 г в фарфоровую чашку. Первые блюда подготовляют к анализу выпариванием до полужидкой или вязкой консистенции. Упаренную массу растирают в фарфоровой ступке до однородного состояния, после чего отбирают навеску в количестве 20 - 30 г. Гарниры подготовляют растиранием в ступке.
Навеску продукта заливают 2 - 3-кратным объемом эфира и переносят с помощью стеклянной палочки и воронки в коническую колбу.
В колбу добавляют для связывания воды безводный карбонат, или сульфат, или гидрофосфат натрия в количестве 12 - 18 г, закрывают ее пробкой и оставляют на 15 - 20 мин для экстракции жира при периодическом взбалтывании содержимого колбы. Жидкую часть фильтруют в стакан. Растворитель отгоняют на водяной бане при температуре 37 - 40 °C (в зависимости от растворителя) и жир досушивают в сушильном шкафу при 102 ± 2 °С 1 ч. Стаканы с оставшимся жиром помещают в холодильник для застывания.
Жир, используемый для поливки блюд, также охлаждают в холодильнике до затвердевания.
Аналогичным образом готовят эталон исходного сливочного масла.
Проведение испытания. Пробы жиров наносят в кювету прибора в виде кружочков диаметром 10 - 15 мм и слоем толщиной 2 - 3 мм так, чтобы испытуемые образцы находились в центре поля зрения смотровой камеры. В качестве контроля рядом с опытными образцами помещают образец сливочного масла. Кювету помещают в смотровую камеру прибора, предварительно подогретого в течение 10 - 15 мин, и наблюдают люминесценцию.
Цвет люминесценции исследуемых образцов сравнивают с цветом люминесценции сливочного масла и дают заключение (табл. 11).
Таблица 11
Показатели люминесценции жиров
|
Вид жира |
Цвет люминесценции |
|
Масло сливочное |
От бледно- до ярко-желтого |
|
Маргарин сливочный |
Голубоватый |
|
Маргарин столовый |
|
|
Маргарин "Любительский" |
|
|
Маргарин "Российский" |
|
|
Маргарин "Экстра" |
|
|
Маргарин особый |
|
|
Кулинарный жир "Украинский" |
Интенсивно-голубой |
|
Кулинарный жир "Белорусский" |
|
|
Сало растительное |
|
2.2.8.2. Определение вида жира в кондитерских кремах, гарнирах, супах и жира, используемого для поливки вторых блюд, по коэффициенту преломления
Метод основан на извлечении жира из продукции растворителем, отгоне растворителя и определении вида жира по коэффициенту преломления.
Аппаратура, материалы, реактивы. Те же, что указаны выше, кроме ЛПК-1, и рефрактометр универсальный типа УРЛ с предельным коэффициентом преломления 1,7 или рефрактометр другой системы, пригодный для определения жира.
Подготовка к испытанию. Подготовку пробы, количество продукта, взятого для определения вида жира, извлечение жира из навески, отгон растворителя и досушивание жира проводят, как указано выше.
Проведение испытания. Расплавленный жир, оставшийся в стакане, с помощью стеклянной палочки с оплавленным концом в количестве 2 капель наносят на призму рефрактометра с температурой 30 - 35 °C (при обязательном термостатировании призм) и отсчитывают коэффициент преломления. Показания рефрактометра приводят к температуре 20 °C.
Одновременно определяют коэффициент преломления сливочного масла, отобранного на производстве и подготовленного к испытанию аналогичным образом.
Коэффициент преломления исследуемого жира сравнивают с коэффициентом преломления сливочного масла и дают заключение.
2.3. Определение сахаров
Определение сахаров проводят, контролируя правильность вложения молока, сахарозы и общего сахара, а также крахмалосодержащих продуктов. В блюдах, изделиях (табл. 12) определяют редуцирующие сахара до инверсии, общее количество редуцирующих сахаров до и после инверсии, сахарозу, а также редуцирующие сахара после гидролиза углеводов (сахара и крахмала).
Таблица 12
Виды сахаров, определяемые в полуфабрикатах, блюдах и кулинарных изделиях
|
Наименование полуфабрикатов, блюд и кулинарных изделий |
Редуцирующие сахара |
Фактическое содержание сахара в блюде (изделии), выраженное в сахарозе |
Редуцирующие сахара после гидролиза крахмала и других углеводов |
Содержание наполнителя в блюде (изделии) |
|
|
до инверсии |
после инверсии (общий сахар) |
||||
|
Полуфабрикаты и изделия из котлетной массы |
- |
- |
- |
+ |
+ |
|
Сельдь рубленая |
- |
+ (сахара яблок и лука) |
- |
+ |
+ |
|
Тефтели |
- |
+ (сахара лука) |
- |
+ |
+ |
|
Мясной фарш с рисом и луком |
- |
+ (сахара лука) |
- |
+ |
+ |
|
Муссы с манной крупой |
- |
+ |
- |
+ |
+ |
|
Творожные полуфабрикаты и изделия |
+ |
+ |
+ |
+ |
+ |
|
Полуфабрикаты из муки (тесто) |
- |
+ |
+ |
- |
|
|
Начинки пирогов и пирожков |
- |
+ |
+* |
+ |
+ |
|
Супы, каши, макаронные, крупяные запеканки, котлеты и биточки из круп, соусы, горячие напитки и другие блюда с молоком (определение лактозы) |
+ |
- |
- |
- |
- |
|
То же, кроме супов, каш и горячих напитков (определение сахарозы) |
+ |
+ |
+ |
- |
- |
|
Молочные кисели, желе (определение лактозы) |
+ |
- |
- |
- |
- |
|
Пудинги, каши, запеченные с фруктами |
- |
+ |
+* |
- |
- |
______________
* Общий сахар, выраженный в сахарозе.
2.3.1. Перманганатный метод Бертрана
Метод основан на способности карбонильных групп сахаров восстанавливать в щелочной среде оксид меди (II) до оксида меди (I). При растворении сульфатом железа (III) аммония образовавшийся оксид меди (I), окисляясь до оксида меди (II), восстанавливает железо (III) в железо (II), количество которого определяют титрованием раствором перманганата калия. Имеется несколько модификаций перманганатного метода, отличающихся концентрацией растворов, продолжительностью окисления и др. и согласно ГОСТам используются для определения редуцирующих сахаров в разных полуфабрикатах и изделиях.
2.3.1.1. Мучные полуфабрикаты (тесто охлажденное), сдобные булочные изделия, сладкие супы, крупяные полуфабрикаты и изделия, сладкие блюда и напитки.
Аппаратура, материалы, реактивы. Бюретки вместимостью 25, 50 см3; колбы для фильтрования под вакуумом вместимостью 500 см3; колбы конические вместимостью 200, 250 см3; насос водоструйный или насос вакуумный Комовского; пипетки на 20, 25 см3; фильтр стеклянный с пластиной № 4 из пористого стекла или фильтры со специально обработанным асбестом, или воронки с асбестом и стеклянным шариком (трубки Аллина); палочки стеклянные; цилиндры вместимостью 25, 50 и 100 см3; плитка электрическая; часы песочные на 3 мин; фарфоровые чашки вместимостью 50 - 100 см3; раствор Фелинга 1, п. 9.4.1; раствор Фелинга 2 (щелочной раствор сегнетовой соли); серная кислота плотностью 1,84 г/см3; раствор перманганата калия концентрации 0,02 моль/дм3 (0,1 н); раствор сульфата аммония железа (III); дистиллированная вода.
Проведение испытания. В коническую колбу вместимостью 200 - 250 см3 вносят пипеткой 20 см3 приготовленного для исследований раствора сахаров (в 20 см3 раствора должно содержаться не более 100 и не менее 10 мг редуцирующих сахаров), приливают из мерного цилиндра по 20 см3 раствора сульфата меди (Фелинг I) и сегнетовой соли (Фелинг II). Смесь осторожно перемешивают, нагревают и кипятят ровно 3 мин с момента образования пузырьков, следя за тем, чтобы кипение не происходило бурно, снимают с огня и дают осадку осесть. Жидкость над осадком должна быть ярко-синей (в случаях обесцвечивания жидкости, что указывает на излишне большую концентрацию сахара в исследуемом растворе, определение следует повторить при большем разведении исследуемого раствора).
По прекращении нагревания выпавшему осадку оксида меди дают осесть, затем фильтруют горячую жидкость через фильтрующую воронку со стеклянным фильтром (или специально приготовленный асбестовый фильтр) в колбу для отсасывания, пользуясь водоструйным или вакуумным насосом для отсасывания жидкости, избегая переноса осадка на фильтр. Как только вся жидкость будет отфильтрована, колбу с осадком и фильтр промывают несколько раз небольшими порциями горячей дистиллированной воды до исчезновения щелочной реакции промывных вод. Осадок оксида меди (I) должен быть все время покрыт жидкостью во избежание соприкосновения его с воздухом и перехода оксида меди (I) в оксид меди (II). Окончив промывание, фильтр вставляют в чистую колбу для отсасывания или оставляют в той же колбе, предварительно освободив и тщательно ополоснув ее от фильтрата и промывных вод. Отмеривают 20 см3 раствора сульфата аммония железа (III), вносят их в коническую колбу с остатком оксида меди и по растворении переносят на фильтр, отсоединив водоструйный насос или насос Комовского. Дают несколько минут постоять для растворения осадка, а затем медленно фильтруют отсасыванием. Колбу и фильтр несколько раз промывают водой до исчезновения кислой реакции, давая каждый раз жидкости стечь с фильтра. Полученный зеленоватый раствор в колбе для отсасывания титруют раствором перманганата калия до появления слабо-розового окрашивания, сохраняющегося в течение 1 мин.
Израсходованное на титрование количество сантиметров
кубических перманганата калия умножают на его титр по меди (![]() ) и по табл. 13 или 14 (в
случае определения лактозы) находят количество инвертного сахара, сахарозы или
лактозы.
) и по табл. 13 или 14 (в
случае определения лактозы) находят количество инвертного сахара, сахарозы или
лактозы.
В мучных полуфабрикатах (тесто) и сдобных булочных изделиях массовую долю общего сахара в сахарозе (Х, %) на сухое вещество рассчитывают по формуле
где:
a - масса сахарозы, найденная по табл. 13, мг;
V - объем исследуемого раствора, приготовленного из навески, см3;
V1 - объем исследуемого раствора, взятый для инверсии сахарозы, см3;
V2 - вместимость мерной колбы, в которой проводилась инверсия, см3;
20 - объем исследуемого раствора, взятый для определения сахаров, см3;
m - масса навески изделия, г;
1000 - пересчет мг в г;
W - влажность изделия.
Массу лактозы (Х1, г на порцию) при определении молока в крупяных изделиях, кофе, какао и редуцирующих сахаров до инверсии сахарозы (Х1, г на порцию) в сладких блюдах рассчитывают по формуле
где:
a - масса лактозы (табл. 14) или редуцирующих сахаров (в инвертном сахаре) до гидролиза сахарозы (табл. 13), мг;
Р - масса блюда (объем напитка), г (см3).
Остальные обозначения, как в формуле (18).
Массу общего сахара (Х2, г на порцию) после инверсии дисахаридов (сахарозы) в кулинарных изделиях и плодово-ягодных напитках находят по формуле
где а2 - масса общего сахара после гидролиза дисахаридов (сахарозы), выраженная в инвертном сахаре.
Остальные обозначения, как в формулах (18) и (19).
Пересчет меди на инвертный сахар или сахарозу, мг
|
Медь |
Инвертный сахар |
Сахароза |
Медь |
Инвертный сахар |
Сахароза |
Медь |
Инвертный сахар |
Сахароза |
|
20,6 |
10 |
9,50 |
79,5 |
41 |
38,95 |
130,8 |
71 |
67,45 |
|
22,6 |
11 |
10,45 |
81,2 |
42 |
39,90 |
132,4 |
72 |
68,40 |
|
24,6 |
12 |
11,40 |
83,0 |
43 |
40,85 |
134,9 |
73 |
69,35 |
|
26,5 |
13 |
12,35 |
84,8 |
44 |
41,80 |
135,6 |
74 |
70,30 |
|
28,5 |
14 |
13,30 |
86,5 |
45 |
42,75 |
137,2 |
75 |
71,25 |
|
30,5 |
15 |
14,25 |
88,3 |
46 |
43,70 |
138,9 |
76 |
72,20 |
|
32,5 |
16 |
15,20 |
90,1 |
47 |
44,65 |
140,5 |
77 |
73,15 |
|
34,5 |
17 |
16,15 |
91,9 |
48 |
45,60 |
142,1 |
78 |
74,10 |
|
36,4 |
18 |
17,10 |
93,6 |
49 |
46,55 |
143,7 |
79 |
75,05 |
|
38,4 |
19 |
18,05 |
95,4 |
50 |
47,50 |
145,3 |
80 |
76, 00 |
|
40,4 |
20 |
19,00 |
97,1 |
51 |
48,45 |
146,9 |
81 |
76, 95 |
|
42,3 |
21 |
19,95 |
98,9 |
52 |
49,40 |
148,5 |
82 |
77,90 |
|
44,2 |
22 |
20,90 |
100,6 |
53 |
50,35 |
150,0 |
83 |
78,85 |
|
46,1 |
23 |
21,85 |
102,3 |
54 |
51,30 |
151,6 |
84 |
79,80 |
|
48,0 |
24 |
22,80 |
104,0 |
55 |
52,25 |
153,2 |
85 |
80,75 |
|
49,8 |
25 |
23,75 |
105,7 |
56 |
53,20 |
154,8 |
86 |
81,70 |
|
51,7 |
26 |
24,70 |
107,4 |
57 |
54,15 |
156,4 |
87 |
82,65 |
|
53,6 |
27 |
25,65 |
109,2 |
58 |
55,10 |
157,9 |
88 |
83,60 |
|
55,5 |
28 |
26,60 |
110,9 |
59 |
56,05 |
159,5 |
89 |
84,55 |
|
57,4 |
29 |
27,55 |
112,6 |
60 |
57,00 |
161,1 |
90 |
85,50 |
|
59,3 |
30 |
28,50 |
114,3 |
61 |
57,95 |
162,6 |
91 |
86, 45 |
|
61,1 |
31 |
29,45 |
115,2 |
62 |
58,90 |
164,2 |
92 |
87,40 |
|
63,0 |
32 |
30,40 |
117,6 |
63 |
59,85 |
165,7 |
93 |
88,35 |
|
64,8 |
33 |
31,35 |
119,2 |
64 |
60,80 |
167,3 |
94 |
89,30 |
|
66,7 |
34 |
32,30 |
120,9 |
65 |
61,75 |
168,8 |
95 |
90,25 |
|
68,5 |
35 |
33,25 |
122,6 |
66 |
62,70 |
170,3 |
96 |
91,20 |
|
70,3 |
36 |
34,20 |
124,2 |
67 |
63,65 |
171,9 |
97 |
92,15 |
|
72,2 |
37 |
35,15 |
125,9 |
68 |
64,60 |
173,4 |
98 |
93,10 |
|
74,0 |
38 |
36,10 |
127,5 |
69 |
65,55 |
175,0 |
99 |
94,05 |
|
75,9 |
39 |
37,05 |
129,2 |
70 |
66,50 |
176,5 |
100 |
95,00 |
|
77,7 |
40 |
38,00 |
|
|
|
|
|
|
Пересчет меди на лактозу, мг
|
Медь |
Лактоза |
Медь |
Лактоза |
|
20 |
11,9 |
105 |
77,1 |
|
25 |
15,6 |
110 |
81,3 |
|
30 |
19,2 |
115 |
85,4 |
|
35 |
22,8 |
120 |
89,6 |
|
40 |
26,5 |
125 |
93,8 |
|
45 |
30,2 |
130 |
98,1 |
|
50 |
33,9 |
135 |
102,4 |
|
55 |
37,7 |
140 |
106,8 |
|
60 |
41,5 |
145 |
111,2 |
|
65 |
45,3 |
150 |
115,6 |
|
70 |
49,2 |
155 |
120,1 |
|
75 |
53,1 |
160 |
124,6 |
|
80 |
57,0 |
165 |
129,2 |
|
85 |
61,0 |
170 |
133,8 |
|
90 |
65,0 |
175 |
138,5 |
|
95 |
69,0 |
180 |
143,8 |
|
100 |
73,0 |
- |
- |
Массу сахарозы (S, г на порцию) в сладких супах и блюдах рассчитывают по разности между количеством общего сахара после гидролиза дисахаридов (сахарозы) Х2 и редуцирующих сахаров до гидролиза дисахаридов (сахарозы) Х1, а в крупяных изделиях, кофе, какао - по разности между массами общего сахара и лактозы Х1 по формуле
|
S = (Х2 - Х1)∙0,95, |
(21) |
где:
X1 - масса редуцирующих сахаров до гидролиза дисахаридов или лактозы, г;
Х2 - масса общего сахара после гидролиза дисахаридов, г;
0,95 - коэффициент пересчета инвертного сахара на сахарозу.
Расхождение между результатами двух параллельных определений в одной лаборатории допускается не более 0,5 %, в разных лабораториях - не более 1 %. За конечный результат испытаний принимают среднее арифметическое двух параллельных определений, вычисленное с точностью до 0,1 %.
2.3.1.2. Мучные кондитерские изделия, полуфабрикаты для тортов и пирожных (ГОСТ 5903-89)
Аппаратура, материалы, реактивы те же, что указаны выше.
Подготовка к испытаниям. 1. Раствор Фелинга 1: 69,28 г сульфата меди растворяют в дистиллированной воде в мерной колбе вместимостью 1 дм3.
2. Раствор Фелинга II: 346 г тартрата калия-натрия растворяют при слабом нагревании в 400 - 500 см3 дистиллированной воды, прибавляют 100 г гидроксида натрия или калия, растворенного в 200 - 300 см3 дистиллированной воды, количественно переносят в мерную колбу вместимостью 1000 см3 и доводят дистиллированной водой до метки.
3. Раствор сульфата аммония железа (III): один объем насыщенного на холоде раствора сульфата аммония железа (III) смешивают с одним объемом серной кислоты, разбавленной 1:10. Раствор квасцов не должен содержать солей оксида железа (II), при прибавлении к раствору одной-двух капель раствора перманганата калия розовая окраска не должна исчезать в течение 1 мин.
4. Раствор перманганата калия: 5 г перманганата калия растворяют в свежепрокипяченной охлажденной дистиллированной воде в мерной колбе вместимостью 1000 см3. Раствор хранят в темной склянке. Через 8 - 14 сут раствор фильтруют через стеклянную вату или асбест. 1 см3 этого раствора соответствует 10 мг меди. Для установления поправочного коэффициента 0,2483 г щавелевой кислоты растворяют в 50 см3 дистиллированной воды, прибавляют 25 см3 серной кислоты, разбавленной 2:5, нагретой до 50 °C на водяной бане, и титруют раствором перманганата калия до розовой окраски. Поправочный коэффициент (K) вычисляют по формуле
|
|
(22) |
где:
V - объем раствора перманганата калия, израсходованный на титрование взятой навески щавелевой кислоты, см3;
25 - объем перманганата калия, соответствующий 0,2483 г щавелевой кислоты, см3.
Проведение испытания. Растворение навески, осаждение несахаров, приготовление фильтрата указано ниже.
В коническую колбу вместимостью 250 см3 вносят пипетками 25 см3 раствора сульфата меди (II), 25 см3 щелочного раствора тартрата калия-натрия и 50 см3 дистиллированной воды. Смесь быстро доводят до кипения, и, не прекращая нагревания, приливают 25 см3 подготовленного раствора исследуемого изделия, и, когда жидкость закипит, кипятят ровно 2 мин.
По прекращении нагревания выпавшему осадку оксида меди дают осесть, затем фильтруют горячую жидкость через фильтрующую воронку со стеклянным фильтром с предварительно нанесенным на него мелковолокнистым асбестом слоем 1 см.
Как только вся жидкость будет отфильтрована, колбы с осадком и фильтр промывают несколько раз небольшими порциями горячей дистиллированной воды. Осадок оксида меди (I) должен быть все время покрыт жидкостью во избежание соприкосновения его с воздухом и перехода оксида меди (I) в оксид меди (II). Окончив промывание, фильтр вставляют в чистую колбу для отсасывания, отмеривают 30 - 50 см3 раствора сульфата аммония железа (III), вносят их в коническую колбу с остатком оксида меди и по растворении переносят на фильтр, отсоединив водоструйный насос или насос Комовского. После растворения всего оксида меди присоединяют водоструйный насос или насос Комовского, колбу и фильтр промывают несколько раз небольшими порциями горячей дистиллированной воды, давая каждый раз жидкости стечь с фильтра (до исчезновения кислой реакции).
Удалив фильтр из колбы для отсасывания, к фильтрату прибавляют 25 - 30 см3 серной кислоты (1:10) и тотчас же титруют раствором перманганата калия до появления слабо-розового окрашивания, не исчезающего в течение 1 мин.
Объем раствора перманганата калия, пошедший на титрование, умножают на 10 и на поправочный коэффициент K, после чего по табл. 15 находят соответствующее количество мг инвертного сахара в 25 см3 раствора исследуемого изделия.
Обработка результатов. Массовую долю общего сахара (Х) в процентах, выраженную в сахарозе, в пересчете на сухое вещество вычисляют по формуле
|
|
(23) |
где:
a - масса инвертного сахара, определенная по табл. 15, мг;
m - масса навески, кг;
V - вместимость мерной колбы, см3;
V1 - объем исследуемого раствора, взятый для анализа, см3;
V2 - вместимость мерной колбы, в которой проводилась инверсия, см3;
V3 - объем раствора, взятый для инверсии, см3;
1000 - коэффициент пересчета инвертного сахара в граммы;
0,95 - коэффициент пересчета инвертного сахара на сахарозу;
W - массовая доля влаги в исследуемом изделии, %.
Результаты параллельных определений вычисляют до второго десятичного знака. Окончательный результат округляют до первого десятичного знака. За результат анализа принимают среднее арифметическое результатов двух параллельных определений, допускаемые расхождения между которыми в одной лаборатории не должны превышать по абсолютной величине 0,5 %, выполненных в разных лабораториях - 1,0 %.
Определение инвертного сахара по массе восстановленной меди, мг
|
Масса меди |
Масса инвертного сахара |
Масса меди |
Масса инвертного сахара |
Масса меди |
Масса инвертного сахара |
Масса меди |
Масса инвертного сахара |
Масса меди |
Масса инвертного сахара |
|
25 |
13,7 |
71 |
37,1 |
116 |
60,7 |
161 |
84,8 |
206 |
109,6 |
|
26 |
14,2 |
72 |
37,5 |
117 |
61,2 |
162 |
85,4 |
207 |
110,2 |
|
27 |
14,7 |
73 |
38,0 |
118 |
61,7 |
163 |
85,9 |
208 |
110,8 |
|
28 |
15,2 |
74 |
38,6 |
119 |
62,3 |
164 |
86,5 |
209 |
111,3 |
|
29 |
15,7 |
75 |
39,1 |
120 |
62,8 |
165 |
87,0 |
210 |
111,9 |
|
30 |
16,2 |
76 |
39,6 |
121 |
63,3 |
166 |
87,6 |
211 |
112,5 |
|
31 |
16,7 |
77 |
40,1 |
122 |
63,9 |
167 |
88,1 |
212 |
113,0 |
|
32 |
17,2 |
78 |
40,6 |
123 |
64,4 |
168 |
88,6 |
213 |
113,6 |
|
33 |
17,7 |
79 |
41,1 |
124 |
64,9 |
169 |
89,2 |
214 |
114,2 |
|
34 |
18,2 |
80 |
41,7 |
125 |
65,5 |
170 |
89,7 |
215 |
114,7 |
|
35 |
18,7 |
81 |
42,2 |
126 |
66,0 |
171 |
90,3 |
216 |
115,3 |
|
36 |
19,2 |
82 |
42,7 |
127 |
66,5 |
172 |
90,8 |
217 |
115,8 |
|
37 |
19,7 |
83 |
43,2 |
128 |
67,1 |
173 |
91,4 |
218 |
116, 4 |
|
38 |
20,2 |
84 |
43,8 |
129 |
67,6 |
174 |
91,9 |
219 |
117,0 |
|
39 |
20,7 |
85 |
44,4 |
130 |
68,1 |
175 |
92,4 |
220 |
117,5 |
|
40 |
21,3 |
86 |
45,0 |
131 |
68,7 |
176 |
93,0 |
221 |
118,1 |
|
41 |
21,8 |
87 |
45,5 |
132 |
69,2 |
177 |
93,5 |
222 |
118,7 |
|
42 |
22,3 |
88 |
45,9 |
133 |
69,7 |
178 |
94,1 |
223 |
119,2 |
|
43 |
22,8 |
89 |
46,4 |
134 |
70,3 |
179 |
94,6 |
224 |
119,8 |
|
44 |
23,3 |
90 |
46,9 |
135 |
70,8 |
180 |
95,2 |
225 |
120,4 |
|
45 |
23,8 |
91 |
47,4 |
136 |
71,3 |
181 |
95,7 |
226 |
120,9 |
|
46 |
24,4 |
92 |
47,9 |
137 |
71,9 |
182 |
96,2 |
227 |
121,5 |
|
47 |
24,9 |
93 |
48,4 |
138 |
72,4 |
183 |
96,8 |
228 |
122,1 |
|
48 |
25,4 |
94 |
48,9 |
139 |
72,9 |
184 |
97,3 |
229 |
122,6 |
|
49 |
25,9 |
95 |
49,5 |
140 |
73,5 |
185 |
97,9 |
230 |
123,2 |
|
50 |
26,4 |
96 |
50,0 |
141 |
74,0 |
186 |
98,4 |
231 |
123,6 |
|
51 |
26,9 |
97 |
50,5 |
142 |
74,5 |
187 |
99,0 |
232 |
124,3 |
|
52 |
27,4 |
98 |
51,1 |
143 |
75,1 |
188 |
99,5 |
233 |
124,9 |
|
53 |
27,9 |
99 |
51,6 |
144 |
75,6 |
189 |
100,1 |
234 |
125,5 |
|
54 |
28,4 |
100 |
52,1 |
145 |
76,1 |
190 |
100,6 |
235 |
126,9 |
|
55 |
28,9 |
101 |
52,7 |
146 |
76,7 |
191 |
101,2 |
236 |
127,0 |
|
56 |
29,5 |
102 |
53,2 |
147 |
77,2 |
192 |
101,7 |
237 |
127,2 |
|
57 |
30,0 |
103 |
53,7 |
148 |
77,8 |
193 |
102,3 |
238 |
127,8 |
|
58 |
30,5 |
104 |
54,3 |
149 |
78,3 |
194 |
102,9 |
239 |
128,3 |
|
59 |
31,1 |
105 |
54,8 |
150 |
78,9 |
195 |
103,4 |
240 |
128,9 |
|
60 |
31,5 |
106 |
55,3 |
151 |
79,4 |
196 |
104,0 |
241 |
129,5 |
|
61 |
32,0 |
107 |
55,9 |
152 |
80,0 |
197 |
104,6 |
242 |
130,0 |
|
62 |
32,5 |
108 |
56,4 |
153 |
80,5 |
198 |
105,1 |
243 |
130,6 |
|
63 |
33,1 |
109 |
56,9 |
154 |
81,1 |
199 |
105,7 |
244 |
131,2 |
|
64 |
33,6 |
110 |
57,5 |
155 |
81,6 |
200 |
106,3 |
245 |
131,8 |
|
65 |
34,1 |
111 |
58,0 |
156 |
82,1 |
201 |
106,8 |
246 |
132,3 |
|
66 |
34,6 |
112 |
58,5 |
157 |
82,7 |
202 |
107,4 |
247 |
132,9 |
|
67 |
35,1 |
113 |
59,1 |
158 |
83,2 |
203 |
107,9 |
248 |
133,5 |
|
68 |
35,6 |
114 |
59,6 |
159 |
83,8 |
204 |
108,6 |
249 |
133,9 |
|
69 |
36,0 |
115 |
60,1 |
160 |
84,3 |
205 |
109,1 |
250 |
134,6 |
|
70 |
36,5 |
|
|
|
|
|
|
|
|
2.3.2. Цианидный метод
Метод основан на способности редуцирующих сахаров восстанавливать в щелочном растворе гексацианоферрат (III) калия в гексацианоферрат (II) калия.
Данный метод применяют для определения количества хлеба в рубленых полуфабрикатах из мяса (птицы, рыбы); риса в фаршах; муки и манной крупы в творожных изделиях; сахарозы в сладких и вторых блюдах, напитках; лактозы в молочных продуктах.
Аппаратура, материалы, реактивы. Колбы конические вместимостью 100 или 150 см3; бюретки для горячего титрования вместимостью 25 или 50 см3 с изогнутым концом или прямые; пипетки вместимостью 5, 10 и 20 см3; капельница стеклянная; часы песочные на 1, 3 мин; плита электрическая; гексацианоферрат (III) калия (красная кровяная соль); феррицианид, раствор массовой долей 1 %; метиленовый голубой; гидроксид натрия, раствор концентрации 2,5 моль/дм3 (2,5 н); вода дистиллированная.
Проведение испытания. Ориентировочное титрование. Бюретку для горячего титрования заполняют испытуемым раствором. В коническую колбу вместимостью 100 см3 вносят 20 см3 раствора гексацианоферрата (III) калия*(6) и 5 мл гидроксида натрия, одну каплю раствора метиленового голубого и доводят до кипения. К непрерывно слабо кипящему раствору приливают из бюретки по каплям (1 капля в сек) испытуемый раствор до первых признаков исчезновения синей окраски, которая при кипении раствора исчезает в течение 3 сек. Появление фиолетовой окраски после остывания раствора во внимание не принимается.
______________
*(6) При концентрации сахара в испытуемом растворе от 0,25 % и выше.
Контрольное титрование. В коническую колбу вместимостью 100 см3 вносят пипеткой 20 или 10 см3 раствора гексацианоферрата (III) калия и соответственно 5 или 2,5 см3 раствора гидроксида натрия, каплю раствора метиленового голубого и из бюретки приливают испытуемый раствор на 0,3 - 0,5 см3 меньше, чем пошло на ориентировочное титрование. Смесь нагревают до кипения в течение 1 - 1,5 мин и, кипятят точно 1 мин при слабом нагреве, затем кипящую жидкость осторожно дотитровывают из бюретки испытуемым раствором до исчезновения синей и появления желтой окраски. Продолжительность кипения не должна превышать 3 мин. По бюретке отсчитывают общее количество испытуемого раствора, пошедшее на титрование. Наиболее точные результаты получаются, когда на титрование уходит 5 - 6 см3 испытуемого раствора.
Обработка результатов. Массовую долю редуцирующих сахаров до инверсии сахарозы (Х) или после гидролиза крахмала (Х1) в процентах (в зависимости от взятого объема раствора гексацианоферрата (III) калия) вычисляют по формуле
где:
K - поправочный коэффициент на раствор гексацианоферрата (III) калия с массовой долей 1 %;
V - объем раствора редуцирующих сахаров, использованный на восстановление 20 или 10 см3 раствора гексацианоферрата (III) калия с массовой долей 1 % при контрольном титровании, см3;
m - масса навески изделия, г;
20,12 и 0,035, 10,06 и 0,0175 - эмпирические коэффициенты;
V1 - объем исследуемого раствора, приготовленного из навески, см3;
1000 - коэффициент пересчета мг в г.
Массовую долю редуцирующих веществ (Х2) после инверсии сахарозы (дисахаридов) в процентах (в зависимости от объема раствора гексацианоферрата (III) калия) вычисляют по формуле
|
|
(26) |
|
|
(27) |
где:
V - объем раствора редуцирующих сахаров, использованный на восстановление 20 или 10 см3 раствора гексацианоферрата (III) калия с массовой долей 1 % при контрольном титровании, см3;
V2 - вместимость мерной колбы, в которой проводилась инверсия сахарозы (дисахаридов), см3;
V3 - объем раствора, взятый для инверсии, см3.
Остальные обозначения, как в формулах (24) и (25).
Содержание сахарозы рассчитывают по формуле (21).
Если содержание сахара выражают в г на порцию, то в формулы вместо числа 100 в числителе ставят Р (масса блюда или изделия, г).
Расхождение между результатами двух параллельных определений не должно превышать 0,5 %. За конечный результат испытаний принимают среднее арифметическое двух параллельных определений, вычисленное с точностью до 0,1 %.
2.3.2.1. Ускоренный цианидный метод
Метод используется для определения содержания лактозы в молоке, молочных супах и напитках с молоком.
Аппаратура, материалы, реактивы. Те же, что и выше, за исключением метиленового голубого, а также сульфата цинка, раствор массовой концентрации 200 г/дм3.
Проведение испытаний. Фильтрат, полученный после осаждения несахаров в пробах молочного супа, напитков с молоком или молока, наливают в количестве 10 - 15 см3 в бюретку для горячего титрования, споласкивают бюретку и сливают его. После этого бюретку вновь заполняют фильтратом.
Ориентировочное титрование. Бюретку вместимостью 25 см3 заполняют испытуемым раствором, предварительно ополоснув ее тем же раствором. В коническую колбу вместимостью 100 см3 вносят 10 см3 раствора гексацианоферрата (III) калия, 2,5 см3 раствора гидроксида натрия. Колбу помещают на плитку с асбестовой сеткой, нагревают до кипения и добавляют 2 см3 сульфата цинка. К слабо кипящей смеси осторожно приливают из бюретки по каплям испытуемый раствор до полного обесцвечивания (переход окраски из желтой в бесцветную). Общая продолжительность кипения должна быть не более 3 мин.
Контрольное титрование. В коническую колбу вносят 10 см3 раствора гексацианоферрата (III) калия, 2,5 см3 раствора гидроксида натрия и вливают из бюретки испытуемый фильтрат в количестве на 0,2 - 0,3 см3 меньше, чем было израсходовано при ориентировочном титровании. Колбу нагревают до кипения в течение 1 мин, кипятят 1 мин, вливают 2 см3 раствора сульфата цинка и, не прекращая кипячения, дотитровывают испытуемым фильтратом до обесцвечивания раствора.
Обработка результатов. Массу лактозы в блюде и напитке в граммах рассчитывают по формуле
где:
0,012 - количество лактозы, необходимое для восстановления 10 см3 раствора гексацианоферрата (III) калия массовой долей точно 1 %;
V - вместимость мерной колбы, в которую перенесена навеска, см3;
Р - масса порции блюда, г, или объем напитка, см3;
K - поправочный коэффициент на объем осадка белка и жира для молочных блюд и напитков: для супов - 0,985; для каши - 0,974; для напитков - 0,996;
V1 - объем фильтрата, пошедший на титрование раствора гексацианоферрата (III) калия с массовой долей точно 1 % (находят умножением объема испытуемого раствора на поправочный коэффициент к титру раствора гексацианоферрата (III) калия с массовой долей 1 %);
m - масса навески блюда, г, или объем напитка, см3.
Массу лактозы в контрольном образце в процентах рассчитывают по формуле
|
|
(29) |
где:
m - масса навески молока, г;
K = 0,996.
Остальные обозначения те же, что и в формуле (28).
Массу лактозы в молочных блюдах и напитках в процентах рассчитывают по формуле
|
|
(30) |
где обозначения те же, что и в формуле (28).
2.3.2.2. Цианидный фотоколориметрический метод
Фотоколориметрический метод с использованием феррицианида калия основан на колориметрировании избытка щелочного раствора феррицианида калия после реакции с редуцирующими сахарами, применяется для всех кондитерских изделий и полуфабрикатов (ГОСТ 5903-89).
Аппаратура, материалы, реактивы. Весы лабораторные; колбы конические вместимостью 100, 1000 см3; пипетки мерные вместимостью 10, 25 см3; плитка электрическая нагревательная; фотоэлектроколориметр, обеспечивающий измерения в интервалах длин волн 315 - 630 нм с основной погрешностью не более 1 % (по коэффициенту пропускания) или 0,1Д (по оптической плотности); часовые стекла диаметром 50 - 60 мм; эксикатор; вода дистиллированная; глюкоза (безводная); гексацианоферрат (III) калия (феррицианид), щелочной раствор; стандартный раствор глюкозы.
Подготовка к испытанию. 1. Щелочной раствор гексацианоферрата (III) калия: взвешивают 8 г гексацианоферрата (III) калия и 28 г гидроксида калия (или 20 г гидроксида натрия). Отдельно растворяют в небольшом количестве дистиллированной воды. Затем оба раствора сливают в мерную колбу вместимостью 1000 см3 и доводят до метки дистиллированной водой. Раствор готов к использованию через сутки. Раствор можно хранить в склянке из темного стекла в течение 2 мес.
2. Стандартный раствор глюкозы: 1,6 г безводной глюкозы взвешивают с точностью до 0,0002 г и растворяют в мерной колбе вместимостью 1000 см3. Предварительно глюкозу выдерживают в эксикаторе над свежепрокаленным хлоридом кальция в течение 3 сут. После растворения навески раствор в колбе доводят до метки. Если раствор готовят на месяц, необходимо внести в колбу 150 г хлорида натрия и хранить в холодильнике.
3. Построение калибровочного графика. В 6 конических колб вместимостью 250 см3 вносят пипеткой по 25 см3 щелочного раствора феррицианида и по 7,0; 7,5; 8,0; 8,5; 9,0; 9,5 см3 стандартного раствора глюкозы. Из бюретки соответственно приливают 9,0; 8,5; 8,0; 7,5; 7,0; 6,5 см3 дистиллированной воды, тем самым доводят объем жидкости в каждой колбе до 41 см3.
Содержимое колбы нагревают до кипения и кипятят в течение 1 мин. Затем охлаждают и измеряют оптическую плотность на фотоэлектроколориметре при 440 нм. Кювету подбирают такого размера, чтобы оптическая плотность была в пределах 0,3 - 0,6 для раствора, содержащего 8,5 см3 глюкозы (на ФЭК-56 и КФК-2 этому соответствует кювета в 10 мм). Оптическую плотность измеряют в каждом растворе не менее трех раз, из полученных данных берут среднее арифметическое значение. По полученным данным строят калибровочный график, откладывая на оси ординат значения оптической плотности, а на оси абсцисс - соответствующие этим значениям массы глюкозы в миллиграммах. Калибровочный график используется для определения редуцирующих веществ и общего сахара.
Проведение испытаний. В коническую колбу вместимостью 100 - 150 см3 вносят пипетками 25 см3 щелочного раствора феррицианида, 10 см3 исследуемого раствора и 6 см3 дистиллированной воды, затем содержимое колбы доводят до кипения, кипятят точно 1 мин.
После охлаждения заполняют раствором кювету и определяют оптическую плотность так же, как и при снятии калибровочного графика. По значению оптической плотности и калибровочному графику определяют соответствующее количество глюкозы.
Если значения оптической плотности будут за пределами 0,3 - 0,6, то анализ повторяют, соответственно изменив количество добавляемого к раствору феррицианида испытуемого раствора.
Обработка результатов. Массовую долю общего сахара в процентах, выраженную в глюкозе, вычисляют по формуле
|
|
(31) |
где:
m - масса навески, г;
a - масса глюкозы, полученная по калибровочному графику, мг;
V - вместимость мерной колбы, см3;
V2 - вместимость мерной колбы, в которой проводилась инверсия, см3;
V3 - объем исследуемого раствора, взятый для инверсии, см3;
V1 - объем исследуемого раствора, взятый для анализа, см3;
1000 - коэффициент пересчета миллиграммов глюкозы в граммы.
Для перерасчета общего сахара, выраженного в глюкозе, в общий сахар, выраженный в сахарозе, полученное значение умножают на коэффициент 0,95.
Массовую долю общего сахара (X1) в процентах, выраженную в сахарозе, в перерасчете на сухое вещество вычисляют по формуле
|
|
(32) |
где:
Х - массовая доля общего сахара, выраженная в глюкозе, %;
W - массовая доля влаги в исследуемом изделии, %.
Расхождение между результатами двух параллельных определений в одной лаборатории допускается не более 0,5 %, в разных лабораториях - не более 1 %. За конечный результат принимают среднее арифметическое двух параллельных определений, вычисленное с точностью до 0,1 %.
2.3.3. Бихроматный метод
Ускоренный метод определения массовой доли общего сахара, применяемый для всех видов кондитерских изделий и полуфабрикатов, не содержащих алкоголь (ГОСТ 5903-89).
Метод основан на окислении всех сахаров сернокислым раствором бихромата калия до углекислоты и воды и колориметрировании образовавшегося иона Cr3+, эквивалентного количеству вступившего в реакцию сахара.
Аппаратура, материалы, реактивы. Баня водяная; весы лабораторные; колбы мерные на 100, 200, 250, 1000 см3; пипетки мерные на 10 см3; стаканы химические вместимостью 50, 100 см3; фотоэлектроколориметр, обеспечивающий измерения в интервалах длин волн 315 - 630 нм с основной погрешностью не более 1 % (по коэффициенту пропускания) или 0,1Д (по оптической плотности); цилиндры мерные на 25, 500 см3; часы песочные на 10 мин или секундомер; эксикатор; вода дистиллированная; бихромата калия сернокислый раствор; кислота серная плотностью 1,84 г/см3; сахароза; фенолфталеин, спиртовой раствор массовой концентрации 10 г/дм3.
Подготовка к испытанию. Готовят сернокислый раствор бихромата калия: 49 г растворяют в 300 см3 дистиллированной воды (I раствор); отдельно к 300 см3 дистиллированной воды осторожно небольшими порциями при перемешивании приливают 300 см3 концентрированной серной кислоты и охлаждают (II раствор). Сначала первый, а затем второй раствор осторожно переливают в мерную колбу вместимостью 1000 см3, охлаждают до комнатной температуры, доводят объем дистиллированной водой до метки. Построение калибровочного графика производят не ранее чем через сутки после приготовления реактива.
Стандартный раствор сахарозы готовят непосредственно перед употреблением: 1,0 г сахарозы или сахара-рафинада, предварительно высушенных в эксикаторе в течение 3 сут, взвешивают с точностью до 0,001 г, растворяют в дистиллированной воде и количественно переносят в мерную колбу вместимостью 250 см3. Объем раствора доводят до метки дистиллированной водой и тщательно перемешивают. Полученный раствор должен содержать 4 мг сахарозы в 1 см3.
Для построения калибровочного графика в 5 мерных колб вместимостью каждая 100 см3 мерным цилиндром вносят по 25 см3 сернокислого раствора бихромата калия, затем пипеткой по 2, 4, 6, 8, 10 см3 стандартного раствора сахарозы и 23, 21, 19, 17, 15 см3 дистиллированной воды, чтобы объем в каждой колбе достиг 50 см3. Колбы с содержимым помещают в кипящую баню на 10 мин, охлаждают до комнатной температуры, доводят объем дистиллированной водой до метки, тщательно перемешивают и измеряют оптическую плотность на ФЭКе при λ = 630 - 670 нм в кювете 30 мм. Оптическую плотность измеряют в каждом растворе не менее 3 раз и из полученных данных берут среднее арифметическое значение. По полученным данным строят калибровочный график, откладывая на оси ординат значения оптической плотности, а на оси абсцисс - соответствующие этим значениям массы сахарозы в мг. Калибровочный график используется для определения общего сахара.
Проведение испытаний. В мерную колбу вместимостью 100 см3 мерным цилиндром вносят 25 см3 сернокислого раствора бихромата калия, 10 см3 фильтрата исследуемого раствора и 15 см3 дистиллированной воды. Колбу помещают в кипящую водяную баню на 10 мин, охлаждают до комнатной температуры, доводят объем дистиллированной водой до метки, тщательно перемешивают и измеряют плотность. По значению оптической плотности и калибровочному графику находят соответствующее количество общего сахара, условно выраженное в сахарозе.
Массовую долю общего сахара (Х) в процентах, выраженную в сахарозе, определяют по формуле
|
|
(33) |
где:
m - масса навески изделия, г;
a - масса сахарозы, полученная по калибровочному графику, мг;
V - вместимость мерной колбы, см3;
V1 - объем исследуемого раствора, взятый для анализа, см3;
1000 - коэффициент пересчета мг сахарозы в г;
K - поправочный коэффициент, учитывающий окисление декстринов (для изделий, содержащих патоку), определяют по табл. 16.
Таблица 16
|
Отношение содержания патоки к содержанию общего сахара, % |
Поправочный коэффициент K |
|
2 - 5 |
0,96 |
|
6 - 10 |
0,94 |
|
11 - 15 |
0,92 |
|
16 - 20 |
0,90 |
|
21 - 30 |
0,88 |
Массовую долю общего сахара (X1) в процентах в пересчете на сухое вещество определяют по формуле
|
|
(34) |
где:
Х - массовая доля общего сахара, выраженная в сахарозе, %;
W - массовая доля влаги в исследуемом изделии, %.
За окончательный результат анализа принимают среднее арифметическое результатов двух параллельных определений, допускаемые расхождения между которыми в одной лаборатории не должны превышать 0,5 %, а выполненных в разных лабораториях - 1 %. Результат вычислений округляют до первого десятичного знака.
2.3.4. Йодометрический метод
Метод основан на восстановлении щелочного раствора меди некоторым количеством раствора редуцирующих веществ и определении количества образовавшегося оксида меди (I) или невосстановившейся меди йодометрическим способом. В качестве щелочного раствора меди используют медно-цитратный раствор. При отсутствии лимонной кислоты, входящей в данный раствор, используют растворы Фелинга 1 и 2 и, соответственно, другую таблицу пересчета количества см3 тиосульфата натрия в мг сахарозы.
Метод применяют для контроля содержания сахара в творожных, мучных полуфабрикатах и изделиях, мучных блюдах и блюдах из творога и др.
Аппаратура, материалы, реактивы. Плитка электрическая; сетка асбестовая; часы песочные на 2 и 10 мин; холодильник шариковый или воздушный с длиной трубки не менее 1 м; колбы конические вместимостью 250, 500 см3; бюретки вместимостью 25, 50 см3; пипетки вместимостью 2, 5, 10, 15 и 25 см3; цилиндры мерные вместимостью 10, 25, 100 см3; йодид калия, раствор с массовой долей 30 %; кислота серная плотностью 1,84 г/см3, раствор концентрации 2 моль/дм3 (4 н), раствор с массовой долей 25 %; натрий серноватистокислый (тиосульфат натрия), раствор концентрации 0,1 моль/дм3 (0,1 н); крахмал растворимый, раствор с массовой долей 1 %; щелочной медно-цитратный раствор (см. ниже); растворы Фелинга 1 и 2 (см. ст. 279); вода дистиллированная.
Проведение испытаний. В коническую колбу вместимостью 250 см3 вносят пипетками 25 см3 щелочного медно-цитратного раствора, 10 см3 подготовленного раствора сахаров, 15 см3 дистиллированной воды и бросают в колбу для равномерного кипения кусочек пемзы или 2 - 3 кусочка керамики. Колбу присоединяют к обратному холодильнику. Раствор в течение 3 - 4 мин доводят до кипения, кипятят точно 10 мин и быстро охлаждают, погружая колбу в холодную проточную воду. В оставшуюся жидкость пипеткой добавляют последовательно 10 см3 раствора йодида калия и 25 см3 раствора серной кислоты концентрацией 2 моль/дм3 (4 н) цилиндром. Серную кислоту доливают осторожно по внутренним стенкам колбы, все время взбалтывая жидкость во избежание выбрасывания ее из колбы за счет выделившегося углекислого газа. После этого тотчас же титруют выделившийся йод 0,1 моль/дм3 (0,1 н) раствором тиосульфата натрия до светло-желтой жидкости. Затем приливают 2 - 3 см3 раствора крахмала и осторожно дотитровывают окрасившуюся в грязно-синий цвет жидкость до появления окраски молочного цвета, приливая в конце титрования по капле раствор тиосульфата натрия.
Контрольный опыт проводят в тех же условиях, для чего берут 25 см3 щелочного медно-цитратного раствора и 25 см3 дистиллированной воды.
Разность между объемом раствора тиосульфата натрия, полученная при контрольном опыте и при определении, умноженная на коэффициент K, соответствует количеству меди, восстановленному редуцирующими веществами, выраженному в см3 точно 0,1 моль/дм3 (0,1 н) раствора тиосульфата натрия, по которому находят количество мг инвертного сахара во взятых 10 см3 раствора навески испытуемого изделия (табл. 17).
Пересчет тиосульфата натрия в инвертный сахар (при использовании растворов Фелинга 1 и 2)
|
Объем точно 0,1 моль/дм3 раствора тиосульфата натрия, см3 |
Массовая доля глюкозы, мг |
|||||||||
|
0 |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
|
|
0 |
0,0 |
0,3 |
0,6 |
1,0 |
1,3 |
1,6 |
1,9 |
2,2 |
2,6 |
2,9 |
|
1 |
3,2 |
3,5 |
3,8 |
4,1 |
4,4 |
4,7 |
5,1 |
5,4 |
5,7 |
6, 0 |
|
2 |
6,3 |
6,6 |
6,9 |
7,2 |
7,5 |
7,8 |
8,2 |
8,5 |
8,8 |
9,1 |
|
3 |
9,4 |
9,7 |
10,0 |
10,4 |
10,7 |
11,0 |
11,3 |
11,6 |
12,0 |
12,3 |
|
4 |
12,6 |
12,9 |
13,3 |
13,6 |
13,9 |
14,2 |
14,6 |
14,9 |
15,2 |
15,6 |
|
5 |
15,9 |
16,2 |
16,6 |
16,9 |
17,2 |
17,5 |
17,9 |
18,2 |
18,5 |
18,9 |
|
6 |
19,2 |
19,5 |
19,9 |
20,2 |
20,5 |
20,8 |
21,2 |
21,5 |
21,8 |
22,1 |
|
7 |
22,4 |
22,7 |
23,0 |
23,4 |
23,7 |
24,0 |
24,3 |
24,6 |
25,0 |
25,3 |
|
8 |
25,6 |
25,9 |
26,3 |
26,6 |
26,9 |
27,2 |
27,6 |
27,9 |
28,2 |
28,6 |
|
9 |
28,9 |
29,2 |
29,6 |
29,9 |
30,3 |
30,6 |
30,9 |
31,3 |
31,6 |
32,0 |
|
10 |
32,3 |
32,6 |
33,0 |
33,3 |
33,7 |
34,0 |
34,3 |
34,7 |
35,0 |
35,4 |
|
11 |
35,7 |
36,0 |
36,4 |
36,7 |
37,0 |
37,3 |
37,7 |
38,0 |
38,2 |
38,7 |
|
12 |
39,0 |
39,3 |
39,7 |
40,0 |
40,4 |
40,7 |
41,0 |
41,4 |
41,7 |
42,1 |
|
13 |
42,4 |
42,7 |
43,1 |
43,4 |
43,8 |
44,1 |
44,4 |
44,8 |
45,1 |
45,5 |
|
14 |
45,8 |
46,1 |
46,5 |
46,8 |
47,2 |
47,5 |
47,9 |
48,2 |
48,6 |
48,9 |
|
15 |
49,3 |
49,6 |
50,0 |
50,3 |
50,7 |
51,0 |
51,4 |
51,7 |
52,1 |
52,4 |
|
16 |
52,8 |
53,1 |
53,5 |
53,8 |
54,2 |
54,5 |
54,9 |
55,2 |
55,6 |
55,9 |
|
17 |
56,3 |
56,6 |
57,0 |
57,3 |
57,7 |
58,0 |
58,4 |
58,7 |
59,1 |
59,4 |
|
18 |
59,8 |
60,1 |
60,5 |
60,8 |
61,2 |
61,5 |
61,9 |
62,2 |
62,6 |
62,9 |
|
19 |
66,3 |
63,7 |
64,0 |
64,4 |
64,7 |
65,1 |
65,5 |
65,8 |
66,2 |
66,5 |
|
20 |
66,9 |
67,3 |
67,7 |
68,0 |
68,4 |
68,8 |
69,2 |
69,6 |
69,9 |
70,3 |
|
21 |
70,7 |
71,1 |
71,5 |
71,8 |
72,2 |
72,6 |
73,0 |
73,4 |
73,7 |
74,1 |
|
22 |
74,5 |
74,9 |
75,3 |
75,7 |
76,1 |
76,5 |
76,9 |
77,3 |
77,7 |
78,1 |
|
23 |
78,5 |
78,9 |
79,3 |
79,7 |
80,1 |
80,5 |
81,0 |
81,4 |
81,8 |
82,2 |
|
24 |
82,5 |
83,0 |
83,4 |
83,8 |
84,2 |
84,6 |
85,0 |
85,4 |
85,8 |
86,2 |
|
25 |
86,6 |
87,0 |
87,4 |
87,8 |
88,2 |
88,6 |
90,0 |
90,4 |
90,8 |
91,2 |
При использовании растворов Фелинга 1 и 2 в коническую колбу вместимостью 200 - 300 см3 отмеривают 10 см3 испытуемого раствора, добавляют по 10 см3 растворов Фелинга 1 и 2, доводят в течение 3 мин до кипения, кипятят ровно 2 мин, быстро охлаждают проточной водой до комнатной температуры, прибавляют 10 см3 раствора йодида калия, 10 см3 раствора серной кислоты с массовой долей 25 % и сейчас же титруют 0,1 моль/дм3 раствором тиосульфата натрия до светло-желтого окрашивания. Затем добавляют 2 см3 раствора крахмала и продолжают титрование до исчезновения синей окраски. Контрольный опыт проводят в тех же условиях, взяв вместо испытуемого раствора 10 см3 дистиллированной воды.
Разность между величинами, полученными при контрольном опыте и при определении сахара в испытуемом растворе, умноженная на поправку к титру, соответствует количеству восстановленной меди, выраженному в см3 точно 0,1 моль/дм3 раствора тиосульфата натрия.
По количеству см3 тиосульфата натрия находят количество мг инвертного сахара во взятых 10 см3 испытуемого раствора (табл. 18).
Пересчет тиосульфата натрия в инвертный сахар (при использовании щелочного медно-цитратного раствора)
|
Объем точно 0,1 моль/дм3 раствора тиосульфата натрия, см3 |
Десятые доли миллилитра |
|||||||||
|
0 |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
|
|
Инвертный сахар, мг |
||||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
|
|
0 |
- |
0,25 |
0,50 |
0,75 |
1,00 |
1,25 |
1,50 |
1,75 |
2,00 |
2,25 |
|
1 |
2,51 |
2,77 |
3,03 |
3,29 |
3,55 |
3,81 |
4,07 |
4,33 |
4,59 |
4,85 |
|
2 |
5,11 |
5,37 |
5,63 |
5,89 |
6,15 |
6,41 |
6,67 |
6,93 |
7,19 |
7,45 |
|
3 |
7,71 |
7,97 |
8,23 |
8,49 |
8,75 |
9,01 |
9,27 |
9,53 |
9,79 |
10,05 |
|
4 |
10,31 |
10,57 |
10,83 |
11,09 |
11,35 |
11,61 |
11,87 |
12,13 |
12,39 |
12,65 |
|
5 |
12,92 |
13,19 |
13,46 |
13,73 |
14,00 |
14,27 |
14,54 |
14,81 |
15,08 |
15,35 |
|
6 |
15,62 |
15,89 |
16,16 |
16,43 |
16,70 |
16,97 |
17,24 |
17,51 |
17,78 |
18,05 |
|
7 |
18,32 |
18,59 |
18,86 |
19,13 |
19,40 |
19,67 |
19,94 |
20,21 |
20,48 |
20,75 |
|
8 |
21,02 |
21,29 |
21,56 |
21,83 |
22,10 |
22,37 |
22,64 |
22,91 |
23,18 |
23,45 |
|
9 |
23,73 |
24,01 |
24,29 |
24,57 |
24,85 |
25,13 |
25,41 |
25,69 |
25,97 |
26,25 |
|
10 |
26,53 |
26,81 |
27,09 |
27,37 |
27,65 |
27,93 |
28,21 |
28,49 |
28,77 |
29,05 |
|
11 |
29,33 |
29,61 |
29,89 |
30,17 |
30,45 |
30,73 |
31,01 |
31,29 |
31,57 |
31,85 |
|
12 |
32,13 |
32,41 |
32,69 |
32,97 |
33,25 |
33,53 |
33,81 |
34,09 |
34,37 |
34,65 |
|
13 |
34,93 |
35,21 |
35,49 |
35,77 |
36,05 |
36,33 |
36,61 |
36,89 |
37,17 |
37,45 |
|
14 |
37,74 |
38,03 |
38,32 |
38,61 |
38,89 |
39,18 |
39,47 |
39,76 |
40,05 |
40,34 |
|
15 |
40,63 |
40,92 |
41,21 |
41,50 |
41,79 |
42,08 |
42,37 |
42,66 |
42,95 |
43,24 |
|
16 |
43,53 |
43,82 |
44,11 |
44,40 |
44,69 |
44,98 |
45,27 |
45,56 |
45,85 |
46,14 |
|
17 |
46,44 |
46,74 |
47,04 |
47,34 |
47,64 |
47,94 |
48,24 |
48,54 |
48,84 |
49,24 |
|
18 |
49,44 |
49,74 |
50,04 |
50,34 |
50,64 |
50,94 |
51,24 |
51,54 |
51,84 |
52,14 |
|
19 |
52,44 |
52,74 |
53,04 |
53,34 |
53,64 |
53,94 |
54,24 |
54,54 |
54,84 |
55,14 |
|
20 |
55,45 |
55,76 |
56,07 |
56,38 |
56,69 |
57,00 |
57,31 |
57,62 |
57,93 |
58,24 |
|
21 |
58,55 |
58,86 |
59,17 |
59,48 |
59,79 |
60,10 |
60,41 |
60,72 |
61,03 |
61,34 |
|
22 |
61,65 |
61,96 |
62,27 |
62,58 |
62,89 |
63,20 |
63,51 |
63,82 |
64,13 |
64,44 |
Обработка результатов испытания. Массовую долю редуцирующего сахара до инверсии или после гидролиза (Х) в процентах, выраженную в инвертном сахаре, рассчитывают по формуле
где:
a - масса инвертного сахара, найденная по табл. 18, мг;
V - объем исследуемого раствора, приготовленного из навески, см3;
m - масса навески изделия (блюда), г;
10 - объем испытуемого раствора, взятый для анализа, см3;
1000 - коэффициент пересчета миллиграммов инвертного сахара в граммы.
Если содержание сахара выражают в г на порцию, то в формулы вместо числа 100 в числителе ставят Р (масса блюда или изделия, г).
Массовую долю общего сахара после инверсии (X1) в процентах, выраженную в инвертном сахаре, рассчитывают по формуле
|
|
(36) |
где:
a1 - масса инвертного сахара, найденная по табл. 18, мг;
V1 - вместимость мерной колбы, в которой проводилась инверсия, см3;
V2 - объем испытуемого раствора, взятый для инверсии, см3.
Остальные обозначения, как в формуле (35).
Для пересчета общего сахара, выраженного в инвертном, в общий сахар, выраженный в сахарозе, его количество умножают на коэффициент 0,95.
Массовую долю сахарозы в процентах или граммах (V2) рассчитывают по формуле
|
Х2 = 0,95∙(Х1 - Х), |
(37) |
где:
Х - массовая доля редуцирующего сахара до инверсии, % или г;
V1 - массовая доля общего сахара после инверсии, % или г.
Расхождение между результатами двух параллельных определений в одной лаборатории допускается не более 0,5 %, в разных лабораториях - не более 1 %. За конечный результат испытаний принимают среднее арифметическое двух параллельных определений, вычисленное с точностью до 0,1 %.
2.3.5. Рефрактометрический метод
Этим методом контролируют содержание сахара в напитках (чай, кофе с сахаром, кофе и какао с молоком), сладких блюдах (кисели плодово-ягодные, молочные, муссы плодово-ягодные, желе, самбуки), в бисквитном и песочном полуфабрикатах, в отделочных полуфабрикатах (некоторое виды кремов). Принцип метода и техника работы с рефрактометром описаны с. 17.
Аппаратура, материалы, реактивы. Рефрактометр типа РПЛ-3, РЛУ, УРЛ-У-4,2, модель 1, ИРФ-457; термометр ртутный стеклянный лабораторный со шкалой до 100 °C с ценой деления 1 °C; весы лабораторные; баня водяная; штатив для пробирок; стакан химический вместимостью 50 - 100 см3; цилиндр измерительный вместимостью 25 см3; палочка стеклянная; пробирки стеклянные; воронка стеклянная диаметром 4 - 5 см; марля; бумага фильтровальная; пипетки вместимостью 10 см3; вода дистиллированная.
Бисквитный и песочный полуфабрикаты*(7). Проведение испытания. В пробирку помещают навеску измельченной пробы полуфабриката массой 2 г, взвешенную с точностью до 0,01 г, прибавляют пипеткой 10 см3 дистиллированной воды с температурой 20 °C. Пробирку закрывают пробкой, энергично встряхивают, затем помещают на 8 мин в водяную баню при 65 - 70 °C. Пробирку периодически встряхивают. Содержимое пробирки охлаждают до 20 °C и фильтруют через складчатый фильтр. Первые две капли фильтрата отбрасывают, последующие одну-две капли наносят на призму рефрактометра, снимают показания и по табл. 19 находят массовую долю сахарозы. Рефрактометрию повторяют 2 - 3 раза. За результат принимают среднее арифметическое значение параллельных определений. Расхождения между параллельными определениями массовой доли сухих веществ одной вытяжки не должны превышать 0,2 %.
______________
*(7) Целесообразно пользоваться методом при производственном контроле.
При концентрации сахара в растворе менее 0,25 % берут 10 см3 раствора гексацианоферрата (III) калия и 2,5 см3 раствора гидроксида натрия.
Массовую долю сахарозы (Х) в процентах в пересчете на сухое вещество рассчитывают по формуле
|
|
(38) |
где:
a - содержание сахарозы в исследуемом изделии, %;
W - влажность изделия, %.
Результат анализа вычисляют с точностью до 0,1 % и сравнивают с расчетным значением, подсчитанным по рецептуре с учетом допустимых отклонений (табл. 54).
Зависимость между коэффициентом преломления и массовой долей сухих веществ и сахарозы
|
Для бисквита |
Для песочного полуфабриката |
||||
|
коэффициент преломления |
массовая доля сухих веществ в фильтрате, % |
массовая доля сахарозы, % |
коэффициент преломления |
массовая доля сухих веществ в фильтрате, % |
массовая доля сахарозы, % |
|
1,3390 |
4,20 |
18,6 |
1,3380 |
3,45 |
14,0 |
|
1,3395 |
4,50 |
20,2 |
1,3382 |
3,60 |
14,5 |
|
1,3400 |
4,90 |
21,7 |
1,3384 |
3,70 |
15,1 |
|
1,3401 |
5,00 |
22,7 |
1,3386 |
3,85 |
15,7 |
|
1,3404 |
5,10 |
23,0 |
1,3388 |
4,00 |
16,2 |
|
1,3405 |
5,15 |
23,3 |
1,3390 |
4,10 |
16, 8 |
|
1,3406 |
5,20 |
23,6 |
1,3400 |
4,80 |
19,6 |
|
1,3407 |
5,30 |
23,9 |
1,3410 |
5,50 |
22,4 |
|
1,3408 |
5,35 |
24,2 |
1,3420 |
6,15 |
26,2 |
|
1,3409 |
5,40 |
24,5 |
1,3430 |
6,80 |
29,0 |
|
1,3410 |
5,50 |
24,8 |
1,3440 |
7,45 |
31,8 |
|
1,3420 |
6,20 |
27,9 |
1,3442 |
7,60 |
32,3 |
|
1,3430 |
6,80 |
31,0 |
1,3444 |
7,74 |
32,9 |
|
1,3440 |
7,45 |
34,4 |
1,3445 |
7,80 |
33,2 |
|
1,3443 |
7,70 |
35,4 |
1,3447 |
7,95 |
33,7 |
|
1,3445 |
7,80 |
35,8 |
1,3449 |
8,05 |
34,3 |
|
|
|
|
1,3470 |
9,45 |
40,0 |
Отделочные полуфабрикаты: кремы "Шарлотт", "Новый", шоколадный "Шарлотт" и др. При растворении навески крема в горячей воде в раствор переходит не только сахар, но и лактоза молока, водорастворимые кислоты, соли. Для приведения данных, получаемых рефрактометрическим методом, в соответствии с данными, получаемыми химическими методами по ГОСТ 5903-89, экспериментально установлены поправочные коэффициенты. Умножая полученный по рефрактометру процент сухих веществ на поправочный коэффициент, получают результат, определяемый химическим методом.
Проведение испытания. В химическом стаканчике вместимостью 50 см3 взвешивают 5 г крема с точностью до 0,01 г, добавляют 20 г дистиллированной воды (60 - 70 °C), перемешивают стеклянной палочкой до однородной массы и сразу же фильтруют через бумажный фильтр.
Фильтрат перемешивают и наносят 1 - 2 капли на измерительную призму рефрактометра. Рефракцию повторяют 2 - 3 раза и рассчитывают среднее арифметическое.
По шкале рефрактометра определяют коэффициент преломления или массовую долю сухих веществ.
Если шкала рефрактометра градуирована на коэффициенты преломления, то по табл. 19 находят массовую долю сухих веществ.
При отсчете показаний прибора необходимо отмечать температуру (с погрешностью не более 1 °C), при которой производят рефракцию. Если температура выше или ниже 20 °C, вводят температурную поправку.
Обработка результатов. Массовую долю водорастворимых сухих веществ (Х) в процентах рассчитывают по формуле
|
|
(39) |
где:
a - отсчет по шкале рефрактометра, %;
m1 - масса раствора с навеской, г;
m - масса навески крема, г.
Массовую долю сахара (сахарозы) в креме (Х1 , %) рассчитывают по формуле
|
X1 = X∙K, |
(40) |
где:
Х - массовая доля водорастворимых сухих веществ, %;
K - коэффициенты пересчета водорастворимых сухих веществ на сахарозу (табл. 20).
Центральной технологической лабораторией общественного питания Минторга Казахстана экспериментально установлена массовая доля водорастворимых сухих веществ для кремов по рефрактометру (табл. 20).
Полученный результат сравнивают с расчетными данными по рецептуре с учетом допускаемых отклонений, которые для крема, согласно ТУ 10.04.08.13-88, составляют минус 1,5 %, плюс 2,0 %.
Пример. Рефрактометрия раствора водной вытяжки крема "Шарлотт" (№ 59) проводилась при температуре 23 °C, a = 8 %. Находим температурную поправку по табл. 3. Она равна 0,22:
8 + 0,22 = 8,22 %; m1 = 25; m = 5 г
![]()
X1 = 0,91∙41 = 37,3 %
(норма 36,4 %)
Заключение: массовая доля сахара в норме.
Показатели массовой доли водорастворимых сухих веществ, общего сахара и коэффициент (K)
|
Наименование |
Массовая доля сухих веществ в водном растворе по рефрактометру, % |
Коэффициент пересчета водораство римых сухих веществ на сахарозу |
Массовая доля общего сахара, % |
|
|
в натуре |
в пересчете на сухое вещество |
|||
|
Кремы: |
40 |
0,91 |
36,4 |
48,5 |
|
"Шарлотт" (основной), рец. 39 (59) |
|
|
|
|
|
Шоколадный "Шарлотт", рец. 45 (67) |
40 |
0,89 |
35,6 |
47,2 |
|
"Новый", рец. 41 (61) |
46,6 |
0,83 |
38,7 |
49,6 |
|
Сливочный (основной), рец. 30 (46) |
51,2 |
0,75 |
38,4 |
44,6 |
|
Сливочный, рец. 30 - 1 (46 - 1), на масле "Любительском" |
51,2 |
0,75 |
38,4 |
45,7 |
|
Сливочный с какао-порошком, рец. 37 (57) |
50,4 |
0,72 |
36,3 |
42,2 |
|
Сливочный шоколадный, рец. 38 (58) |
41,2 |
0,67 |
27,6 |
36,3 |
|
"Новый", рец. 41 - 1 (61 - 1), на масле "Любительском" |
46,6 |
0,81 |
40,5 |
51,9 |
|
"Новый" шоколадный, рец. 42 - 1 (62 - 1), на масле "Любительском" |
48,8 |
0,81 |
39,5 |
50,2 |
|
Суфле, рец. 62 (105) |
60,1 |
0,75 |
45,1 |
59,3 |
|
Суфле шоколадное, рец. 63 (106) |
51,6 |
0,84 |
43,4 |
54,9 |
2.3.6. Определение содержания сахара в пересчете на водную фазу в креме (полуфабрикате) для мучных кондитерских изделий*(8)
_______________
*(8) Санитарные правила для предприятий и цехов, вырабатывающих кондитерские изделия с кремом, № 227780 от 18.12.80.
Массовую долю сахара в креме (отделочном полуфабрикате для мучных кондитерских изделий) рассчитывают в процентах в пересчете на сухое вещество. Однако для санитарного контроля при исследовании кремов расчет содержания сахара в них целесообразно проводить в пересчете на водную фазу (воду, содержащуюся в продуктах, входящих в крем по рецептуре). Такой расчет необходим в связи с тем, что кремы являются хорошей средой для размножения стафилококков и накопления их энтеротоксинов и могут быть причиной пищевых отравлений, если в них нет достаточной концентрации сахара. Известно, что сахар повышает осмотическое давление в среде и тем самым прекращает жизнедеятельность микроорганизмов. Содержание сахара в водной фазе и влажность крема находятся в обратной зависимости, чем выше влажность крема, тем ниже концентрация сахара в водной фазе.
В литературе имеются данные, что при содержании сахара в водной фазе в количестве 60 % и более он оказывает задерживающее действие на их развитие.
Для проведения расчета содержания сахара в креме на водную фазу необходимо предварительно определить содержание сахара в натуре. Это определение производится по формуле
|
|
(41) |
где:
С - содержание сахара в натуре, %;
А - содержание сахара, % на сухое вещество, определенное экспериментально;
В - влажность крема, %.
Расчет сахара на водную фазу производится по формуле
где:
Kв - концентрация сахара в водной фазе;
В - влажность крема, %;
С - содержание сахара в натуре, %.
Пример. По данным лабораторного анализа, влажность крема В = 25 %, содержание сахара на сухое вещество А = 51,6 %. Следовательно, в 100 г сухого вещества крема содержится 51,6 % сахара. При расчете по формуле (42) массовая доля сахара в креме (в натуре) с влажностью 25 % составляет 38,7 %. Таким образом, в 100 г крема содержится: воды - 25 г, сахара - 38,6 г, что в сумме составляет 63,6 г (водяная фаза). Содержание сахара на водную фазу составит:
![]()
Заключение: массовая доля сахара в водной фазе крема в норме.
2.4. Определение крахмала
Содержание крахмала определяют при контроле качества полуфабрикатов и готовых изделий, в рецептуру которых входят крахмалсодержащие продукты (хлеб, крупа, пшеничная мука). Крахмал продукта гидролизуют соляной кислотой до глюкозы, определяют содержание последней и пересчитывают ее на крахмал.
2.4.1. Определение хлеба
Правильность вложения хлеба в полуфабрикаты и кулинарные изделия из рубленого мяса, рыбы и птицы без добавления лука определяют в соответствии с ГОСТ 4288-76.
Аппаратура, материалы, реактивы. Весы лабораторные; плитка электрическая; сетка асбестовая; холодильник стеклянный лабораторный; стаканы химические или чашки фарфоровые вместимостью 25, 50 см3; колбы конические вместимостью 100, 250 см3; цилиндры мерные вместимостью 5 и 10 см3; колбы мерные вместимостью 250 см3; воронки стеклянные; бумага фильтровальная; кислота соляная, раствор концентрации 100 г/дм3; гидроксид натрия, раствор концентрации 150 г/дм3; гексацианоферрат (II) калия, раствор концентрации 150 г/дм3; сульфат цинка 7-водный, раствор концентрации 300 г/дм3; метиловый красный, раствор концентрации 1 г/дм3; вода дистиллированная.
Проведение испытания. От пробы берут навеску массой 5 г с точностью до 0,01 г в химический стакан или фарфоровую чашку и добавляют 10 см3 дистиллированной воды. Содержимое стакана (чашки) размешивают стеклянной палочкой и переносят в коническую колбу вместимостью 250 см3, смывая частицы, прилипшие к стакану (чашке) и палочке. Общее количество воды не должно превышать 40 см3.
В колбу с навеской добавляют 30 - 35 см3 раствора соляной кислоты, присоединяют к водному или воздушному холодильнику, ставят на плитку, подложив под колбу асбестовую сетку, и кипятят в течение 10 мин, считая время с момента закипания содержимого колбы. После 10 мин кипячения колбу снимают с плитки, охлаждают струей холодной воды до комнатной температуры. Полученный гидролизат нейтрализуют до слабокислой реакции раствором гидроксида натрия или калия, используя в качестве индикатора каплю раствора метилового красного.
Содержимое колбы после нейтрализации количественно переносят в мерную колбу вместимостью 250 см3, смывая прилипшие к стенкам частицы. Для осветления гидролизата и осаждения белков добавляют пипеткой 3 см3 раствора гексацианоферрата (II) калия и 3 см3 раствора сульфата цинка, доводят дистиллированной водой до метки, тщательно взбалтывают, дают осадку осесть, после чего фильтруют через сухой складчатый фильтр в сухую колбу.
В полученном растворе гидролизата определяют массовую долю редуцирующего сахара, образующегося при гидролизе крахмала, йодометрическим, цианидным методами.
Массовую долю хлеба (У, %) рассчитывают по формуле
где:
Х - массовая доля редуцирующих сахаров, %;
0,9 - коэффициент пересчета глюкозы на крахмал;
48 - коэффициент пересчета крахмала на хлеб.
Если содержание хлеба в исследуемом объекте нужно выразить в г, то вместо 100 ставят массу полуфабриката или кулинарного изделия.
Правильность вложения хлеба в полуфабрикаты и кулинарные изделия из рубленого мяса и рыбы с добавлением лука (тефтели, фрикадельки и др.) определяют с учетом количества редуцирующих сахаров, образующихся из дисахаридов сырья. Для этого гидролиз проводят дважды: при жестком режиме, когда гидролизуются крахмал и дисахариды, и при более мягком, когда гидролизуются только дисахариды.
Аппаратура, материалы, реактивы. Колбы мерные вместимостью 100 см3; баня водяная; соляная кислота, раствор с массовой долей 20 %; кристаллическая сода; остальное, как на с. 74.
Проведение испытания. Навеску исследуемой пробы массой 15 - 25 г переносят в мерную колбу вместимостью 250 см3 с помощью 100 - 150 см3 дистиллированной воды. Содержимое колбы настаивают в течение 20 - 25 мин при частом взбалтывании для извлечения растворимых углеводов. Для осаждения несахаров добавляют 5 см3 раствора гексацианоферрата (II) калия и 5 см3 раствора сульфата цинка. Содержимое колбы доводят до метки дистиллированной водой, перемешивают, дают осадку отстояться и фильтруют через складчатый фильтр в сухую колбу.
Для гидролиза дисахаридов 50 см3 фильтрата пипеткой переносят в мерную колбу вместимостью 100 см3, добавляют 5 см3 раствора соляной кислоты с массовой долей 20 %. В колбу опускают термометр, помещают ее в водяную баню и нагревают в течение 10 мин при температуре 70 °C. Затем содержимое колбы быстро охлаждают до комнатной температуры и нейтрализуют кристаллической содой или раствором гидроксида натрия в присутствии метилового красного до слабокислой реакции. Раствор в колбе доводят до метки дистиллированной водой и перемешивают. В растворе определяют количество редуцирующих сахаров йодометрическим или цианидным методами (с. 55, 64).
Общее количество редуцирующих сахаров после гидролиза крахмала определяют, как на с. 75.
Массовую долю хлеба (У, %) в полуфабрикатах и изделиях, приготовленных с пшеничным хлебом из муки 1 с, за исключением батонов нарезных и городских булочек, рассчитывают по формуле
|
|
(44) |
где
Х3 - массовая доля редуцирующих сахаров после гидролиза крахмала, %;
Х2 - массовая доля редуцирующих сахаров после гидролиза дисахаридов.
Остальные обозначения, как в формуле (43).
Массовую долю хлеба (У1, %) в полуфабрикатах и изделиях, приготовленных с использованием батонов нарезных и городских булочек, рассчитывают по формуле
|
|
(45) |
где a - массовая доля моно- и дисахаридов, добавленных с хлебом, %.
Остальные обозначения, как в предыдущей формуле.
2.4.2. Определение риса
Правильность вложения риса определяют при контроле качества полуфабрикатов и кулинарных изделий, для приготовления которых используют фарш с рисом: голубцов, кабачков, баклажанов, перца или помидоров, фаршированных мясом и рисом, пирожков.
Определение крахмала производят, как указано выше, с увеличением времени гидролиза до 30 мин.
Массу риса в фарше (У, г на порцию) рассчитывают по формуле
где:
Х - массовая доля редуцирующих сахаров, %;
0,9 - коэффициент пересчета на крахмал;
Р - масса блюда, г;
77,3 - содержание углеводов в рисе, %.
2.4.3. Определение манной крупы и пшеничной муки
Правильность вложения манной крупы контролируют в изделиях из творога и муссах, муки - в изделиях из творога.
Аппаратура, материалы, реактивы. Те же, что указаны на с. 74, а также сульфат цинка, раствор концентрации 150 г/дм3 вместо 30-%-ного р-ра.
Проведение испытаний. Для определения крахмала навеску 2 - 4 г изделия из творога или 10 г мусса на манной крупе количественно переносят в коническую колбу вместимостью 250 см3, смывая частицы 80 - 100 см3 теплой дистиллированной воды (50 - 60 °C), добавляют 30 см3 раствора соляной кислоты массовой долей 10 %, присоединяют к холодильнику и нагревают до кипения. Кипятят в течение 30 мин. Далее поступают так, как указано на с. 75. В полученном фильтрате определяют общее содержание редуцирующих сахаров цианидным методом (с. 55).
Массу манной крупы или муки (У, г на порцию) рассчитывают по формуле
|
|
(47) |
где:
X3 - массовая доля редуцирующих сахаров после гидролиза крахмала (общее содержание редуцирующих сахаров), %;
Х2 - массовая доля редуцирующих сахаров после гидролиза дисахаридов, %;
a - массовая доля крахмала, %, в манной крупе 70,3; пшеничной муке 1-го сорта 67,1.
Остальные обозначения, как в формуле (46).
2.5. Методы контроля свежести сырья, полуфабрикатов и готовых блюд и изделий
2.5.1. Определение общей (титруемой) кислотности
Метод применяется для определения степени свежести, а также соответствия кислотности, установленной в полуфабрикатах и кулинарных изделиях (прил. 1).
Под общей кислотностью подразумевается содержание в продукте всех кислот и их кислых солей, реагирующих со щелочью при титровании в присутствии фенолфталеина. Выражают кислотность в градусах или процентах какой-либо кислоты.
В полуфабрикатах из муки, булочных изделиях кислотность измеряют в градусах кислотности, в молочных продуктах - в градусах Тернера (°Т). За градусы титруемой кислотности принимают количество кубических сантиметров раствора гидроксида натрия (гидроксида калия) концентрацией 1 моль/дм3 (1 н), необходимое для нейтрализации кислот, содержащихся в 100 г продукта. За градусы Тернера принимают количество кубических сантиметров гидроксида натрия (гидроксида калия) концентрацией 0,1 моль/дм3, необходимое для нейтрализации кислот, содержащихся в 100 см3 или 100 г продукта.
Аппаратура, материалы, реактивы. Весы лабораторные; вата медицинская гигроскопическая; воронки стеклянные; бюретки вместимостью 25 см3, 50 см3; капельницы; колбы конические вместимостью 100, 250 и 500 см3; палочки стеклянные; пестики 1, 2 или 3; пипетки вместимостью 50 см3; стаканы химические вместимостью 50, 100, 250 см3; ступки 4, 5 или 6; термометр с диапазоном измерения 0 - 150 °C с ценой деления не более 2 °C; цилиндры мерные 100, 250 см3; бумага лакмусовая синяя; бумага фильтровальная; вода дистиллированная; гидроксид натрия или гидроксид калия; растворы концентрации 0,1 моль/дм3 (0,1 н) или стандарт-титры (фиксаналы) в ампулах концентрации 0,1 моль/дм3 (0,1 н); фенолфталеин, спиртовой раствор массовой концентрации 10 г/дм3; тимолфталеин, спиртовой раствор массовой концентрации 1 г/дм3.
Проведение испытания. Навеску измельченной пробы мучных кулинарных, мучных кондитерских, булочных изделий (25 г), взвешенную с точностью 0,01 г, без потерь переносят в коническую колбу 500 см3. Мерную колбу вместимостью 250 см3 наполняют до метки дистиллированной водой и сливают четвертую часть воды в колбу с навеской. Навеску быстро растирают стеклянной палочкой с резиновым наконечником до получения однородной массы, после чего приливают всю оставшуюся воду, закрывают колбу пробкой, энергично встряхивают в течение 2 мин и оставляют при комнатной температуре на 10 мин. Затем смесь снова энергично встряхивают 2 мин и оставляют еще на 8 мин.
Вытяжку из вышеуказанных изделий можно приготовить ускоренным методом. Для этого к навеске добавляют всю воду (250 см3), нагретую до 60 - 70 °C, и встряхивают жидкость 3 мин, после чего оставляют колбу на 1 мин. Отстоявшийся жидкий слой фильтруют в стакан или коническую колбу через вату, марлю или фильтровальную бумагу. Затем в две конические колбы отмеривают пипеткой по 50 см3 фильтрата.
Навеску (5 г) из муки (дрожжевое или дрожжевое слоеное тесто) помещают в ступку и растирают с 50 см3 воды до образования однородной массы. Болтушку переносят в сухую коническую колбу.
Навеску творожных полуфабрикатов и кулинарных изделий (5 г) отвешивают в стеклянный стакан, добавляют 50 см3 воды, нагретой до 35 - 40 °C, и тщательно растирают стеклянной палочкой с резиновым наконечником.
Напитки и сиропы фильтруют. Затем в три конические колбы вместимостью 250 см3 мерным цилиндром наливают по 100 см3 дистиллированной воды, освобожденной от двуокиси углерода. От средней пробы напитка отбирают пипеткой по 10 см3 в каждую из колб (темноокрашенные напитки берут в количестве 5 см3). Сиропы отбирают пипеткой по 2 см2 в колбы с 200 см2 дистиллированной воды.
К полученным фильтратам (болтушкам) добавляют 2 - 3 капли спиртового раствора фенолфталеина и титруют 0,1 моль/дм3 (0,1 н) раствором щелочи до слабо-розового окрашивания, не исчезающего в течение 1 мин.
Одну из колб с напитком, разведенным водой, используют при титровании для сравнения окраски титруемого раствора с первоначальной.
Интенсивно окрашенные фильтраты титруют, используя в качестве индикатора спиртовой раствор тимолфталеина. Конец титрования устанавливают по появлению синей окраски, не исчезающей в течение 1 мин.
Можно титровать окрашенные фильтраты, применяя в качестве индикаторов синюю лакмусовую бумагу. По мере титрования капли титруемой жидкости наносят при помощи стеклянной палочки на полоску лакмусовой бумаги. Титруют до исчезновения покраснения. Чтобы лучше уловить исчезновение красной окраски на лакмусовой бумаге, следует под конец титрования рядом с каплей испытуемой жидкости нанести каплю дистиллированной воды для сравнения и кончать титрование, когда не будет заметно разницы в оттенках двух капель.
Проводят не менее двух параллельных определений.
Кислотность булочных, мучных кондитерских и кулинарных изделий (Х, град.) рассчитывают по формуле
где:
K - поправочный коэффициент гидроксида натрия или калия концентрацией 0,1 моль/дм3;
V - объем раствора гидроксида натрия или калия, израсходованный на титрование;
V1 - объем дистиллированной воды, взятый для растворения навески, см3;
100 - коэффициент пересчета на 100 г продукта;
V2 - объем фильтрата, взятый для титрования, см3;
m - масса навески, г;
10 - коэффициент перерасчета раствора гидроксида натрия или калия концентрации 0,1 моль/дм3 в 1 моль/дм3.
Допустимое расхождение между результатами параллельных определений не должно превышать 0,2°. За конечный результат принимают среднее арифметическое двух параллельных определений, вычисленное с точностью до 0,1°.
Если кислотность требуется выразить в процентах какой-либо кислоты, то градусы кислотности умножают на соответствующий миллиэквивалент, который равен для кислот: уксусной - 0,060; молочной - 0,090; яблочной - 0,067; лимонной - 0,070; винной - 0,075.
Кислотность полуфабрикатов из муки (теста) рассчитывают по формуле
|
Х = 2∙V∙K, |
(49) |
где 2 - коэффициент для пересчета результатов титрования в градусы.
Остальные обозначения, как в предыдущей формуле.
Кислотность творожных полуфабрикатов (изделий) рассчитывают по формуле
|
Х = 20∙V∙K, |
(50) |
где:
Х - кислотность в градусах Тернера (°Т);
20 - коэффициент пересчета результатов титрования в градусы Тернера.
Остальные обозначения, как в формуле (48).
Расхождение между результатами параллельных определений не должно превышать 2 °Т. За конечный результат анализа принимают среднее арифметическое, вычисленное с точностью до 0,1°.
Кислотность напитков и сиропов (Х, см3) выражают количеством кубических сантиметров раствора гидроксида натрия или калия концентрацией 1 моль/дм3, израсходованного на титрование 100 см3 напитка (сиропа), и рассчитывают по формуле
|
|
(51) |
где:
V - объем раствора гидроксида натрия или калия концентрацией 0,1 моль/дм3, израсходованный на титрование;
K - поправочный коэффициент раствора гидроксида натрия или калия концентрацией 0,1 моль/дм3;
V1 - объем напитка (сиропа), взятый на определение, см3.
Вычисление проводят до 0,01 см3 с последующим округлением до 0,1 см3.
2.5.2. Определение активной кислотности
Активная кислотность является показателем качества такой кулинарной продукции, как бульоны, мясные полуфабрикаты и охлажденные блюда.
Определяют активную кислотность электрометрически с помощью pH-метров разных марок по ГОСТ 8756.16-70 "Продукты пищевые консервированные. Методы определения активной кислотности". Метод основан на измерении водородных ионов в испытуемом растворе.
Подготовка к испытанию. Бульоны фильтруют через бумажный складчатый фильтр в сухую колбу.
Из натуральных мясных полуфабрикатов готовят водную вытяжку. Для этого из разных мест исследуемого образца мяса (без жира и соединительной ткани) отбирают навеску массой 10 г, нарезают ее на 30 - 40 кусочков и помещают в коническую колбу вместимостью 250 см3. Затем в колбу наливают 100 см3 предварительно прокипяченной и охлажденной дистиллированной воды и настаивают мясо в течение 15 мин, периодически встряхивая колбу. Полученную вытяжку фильтруют через бумажный складчатый фильтр в сухую колбу. Определение проводят по ГОСТ 8756.16-70.
2.5.3. Определение щелочности
Содержание щелочей регламентируется в песочном тесте, выпеченных из него полуфабрикатах для тортов и пирожных, а также других мучных изделиях, изготовляемых с применением химических разрыхлителей.
Определение щелочности методом титрования основано на нейтрализации щелочных веществ, содержащихся в навеске, кислотой в присутствии бромтимолового синего до появления желтой окраски. Выражают щелочность в градусах. За градусы титруемой щелочности принимают количество кубических сантиметров раствора соляной кислоты (серной кислоты) концентрацией 1 моль/дм3 (1 н), необходимое для нейтрализации щелочных веществ, содержащихся в 100 г продукта.
Аппаратура, материалы, реактивы. Весы лабораторные; воронки стеклянные; капельница; колбы конические вместимостью 250 или 500 см3; бумага фильтровальная лабораторная; бутылки (типа молочной) вместимостью 500 см3; бюретки вместимостью 25 или 50 см3; вата медицинская гигроскопическая; марля медицинская; палочки стеклянные; пипетки вместимостью 50 см3; пестики 1, 2 или 3; стаканы химические вместимостью 50, 250 см3; ступки 4, 5 или 6; цилиндр мерный вместимостью 250 см3; бромтимоловый синий (1 г растворяют в 100 см3 этилового спирта); вода дистиллированная; кислота серная, раствор концентрации С (1/2H2SO4) = 0,1 моль/дм3 (0,1 н); кислота соляная, раствор концентрации 0,1 моль/дм3 (0,1 н).
Проведение испытания. Навеску измельченной пробы (25 г), взвешенную с точностью до 0,01 г, помещают без потерь в сухую бутылку (типа молочной) вместимостью 500 см3, вливают 250 см3 дистиллированной воды, закрывают пробкой и тщательно перемешивают взбалтыванием. Содержимое колбы оставляют на 30 мин, взбалтывая каждые 10 мин, затем фильтруют через вату, или два слоя марли, или фильтровальную бумагу в сухую колбу или стакан. Пипеткой вносят 50 см3 фильтрата в коническую колбу вместимостью 250 см3, прибавляют 2 - 3 капли бромтимолового синего и титруют раствором серной или соляной кислоты до появления желтого окрашивания.
Щелочность Х (в градусах) вычисляют по формуле
|
|
(52) |
где:
К - поправочный коэффициент 0,1 моль/дм3 раствора соляной или серной кислоты, использованного для титрования;
V - объем раствора серной или соляной кислоты, израсходованный на титрование, см3;
V1 - объем дистиллированной воды, взятый для растворения навески, см3;
100 - коэффициент перерасчета на 100 г продукта;
V2 - объем фильтрата, взятый для титрования, см3;
m - масса навески, г;
10 - коэффициент пересчета 0,1 моль/дм3 раствора серной или соляной кислоты в 1 моль/дм3.
Щелочность вычисляют с точностью до 0,1 градуса. Расхождения между параллельными определениями, выполненными в одной лаборатории, не должны превышать 0,2 град., выполненных в разных лабораториях - 0,3 градуса.
2.6. Определение белков
2.6.1. Метод Къельдаля (арбитражный)
Предназначен для определения содержания белка в блюде или рационе с целью контроля его энергетической ценности. Сущность метода состоит в разрушении органического вещества навески концентрированной серной кислотой в присутствии катализаторов (сульфата меди (II) и сульфата натрия). Выделившийся в результате реакции азот улавливается серной кислотой, и образуется сульфат аммония. При добавлении едкого натра выделяется аммиак, который отгоняют в раствор серной кислоты и оттитровывают. Для пересчета на содержание белка количество азота, содержащегося в навеске, умножают на соответствующий коэффициент. Коэффициенты пересчета выведены на основании процентного содержания азота в отдельных видах продуктов.
Аппаратура, материалы, реактивы. Весы лабораторные; плитка электрическая или газовые горелки; сетка асбестовая; колбы Къельдаля вместимостью 100, 200 см3 с грушевидной стеклянной пробкой; цилиндры измерительные вместимостью 10, 25, 50, 100, 250 см3; штатив; колба плоскодонная вместимостью 500 см3 и 1 дм3; колба мерная вместимостью 1 дм3; каплеуловитель стеклянный лабораторный; холодильник стеклянный лабораторный; трубки резиновые; пробки резиновые; трубка стеклянная; капельница стеклянная лабораторная; воронка стеклянная диаметром 5 - 7 см; наконечник стеклянный; бюретка вместимостью 25 см3; колба коническая вместимостью 200 см3; фольга алюминиевая; бумага лакмусовая (красная и синяя); фильтры обеззоленные; палочка стеклянная; серная кислота плотностью 1,84 г/см3; сульфат меди (II); сульфат натрия; гидроксид натрия, раствор с массовой долей 33 %; фиксанал серной кислоты, 0,05 моль/дм3 (0,1 н) раствор; фиксанал гидроксида натрия, 0,1 моль/дм3 (0,1 н) раствор (поправочный коэффициент устанавливают по 0,05 моль/дм3 (0,1 н) раствору серной кислоты); фенолфталеин, спиртовой раствор с массовой долей 1 %; спирт этиловый; вода дистиллированная; метиленовый голубой, водный раствор с массовой долей 0,1 % (раствор 1); метиловый красный, спиртовой раствор с массовой долей 0,02 % (раствор 2); смешанный индикатор (к 25 см3 раствора метиленового голубого с массовой долей 0,1 % добавляют 3 см3 спиртового раствора метилового красного с массовой долей 0,02 %); лакмусовая бумага.
Проведение испытания. На взвешенный обеззоленный фильтр размером 3×3 см берут навеску с точностью 0,001 г с таким расчетом, чтобы в пробе содержалось примерно 20 - 25 мг азота (для продуктов животного происхождения - 0,5 - 1,0 г, для продуктов растительного происхождения, за исключением блюд из бобовых, - свыше 1 г). Очень влажные навески взвешивают в лодочке из фольги или в обрезанной пробирке. Навеску вместе с фильтровальной бумагой, фольгой или пробиркой помещают в колбу Къельдаля вместимостью 100 см3.
При помощи измерительного цилиндра в колбу приливают (соответственно взятой навеске) 10 - 20 см3 концентрированной серной кислоты плотностью 1,84 г/см3. Туда же вводят катализаторы: 0,5 г сульфата меди (II) и 7,5 г сульфата натрия. Колбу устанавливают в наклонном положении в вытяжном шкафу с помощью железного штатива на электрическую плитку с асбестовой сеткой и приливают 1 см3 этилового спирта во избежание выбрасывания жидкости. Колбу закрывают специальной стеклянной пробкой, вначале осторожно подогревают и производят сжигание в течение 4 - 8 ч в зависимости от состава исследуемого блюда. При вспенивании колбу снимают с огня и слегка встряхивают для разрушения пены. Сжигание можно считать законченным, когда содержимое колбы станет прозрачным, бесцветным или слегка зеленоватым.
Далее собирают прибор для отгона аммиака. Плоскодонную колбу с помощью резиновой пробки соединяют через насадку-каплеуловитель с концом шарикового холодильника; холодильник закрепляют на штативе и подводят к нему воду. В эту же пробку вставляют стеклянную трубку, на которую надевают резиновую трубку с зажимом*(9). К нижнему концу холодильника через резиновую трубку присоединяют стеклянный наконечник. Наконечник опускают в коническую колбу вместимостью 250 см3, куда предварительно приливают 40 см3 0,05 моль/дм3 (0,1 н) раствора серной кислоты для улавливания аммиака. Наконечник погружают на 1,5 - 2 см в раствор серной кислоты*(10), проверяют герметичность соединений.
Содержимое колбы Къельдаля слегка охлаждают*(11), в колбу приливают осторожно по стенке около 50 см3 дистиллированной воды, жидкость перемешивают круговыми движениями и переливают в плоскодонную колбу вместимостью 0,5 - 1 дм3. Колбу Къельдаля несколько раз ополаскивают дистиллированной водой, промывные воды переносят в ту же плоскодонную колбу. Всего на перенесение навески используют 150 - 200 см3 дистиллированной воды.
________________
*(9) Это приспособление используют для добавления раствора щелочи с массовой долей 33 % в колбу с минерализованной навеской при полной герметичности прибора.
*(10) В противном случае выделяющийся в момент внесения щелочи свободный аммиак в первые 15 мин после начала отгонки не свяжется с титрованной кислотой, следовательно, не сможет быть учтен.
*(11) При полном охлаждении минерализата выпадают сернокислые соли, которые при разбавлении водой растворяются очень медленно.
В плоскодонную колбу, куда помещен раствор, опускают красную лакмусовую бумагу и вливают раствор гидроксида натрия с массовой долей 33 % при помощи воронки, вставленной в приспособление для вливания щелочи, из расчета 40 см3 на каждый 10 см3 серной кислоты, взятой для сжигания навески. Вливание раствора щелочи производят до тех пор, пока лакмусовая бумага не станет отчетливо синего цвета. Вливание щелочи следует производить очень осторожно, чтобы не произошло выбрасывания содержимого колбы.
После вливания раствора щелочи систему для отгонки плотно присоединяют к колбе и еще раз проверяют на герметичность. Содержимое плоскодонной колбы перемешивают круговыми движениями. Колбу укрепляют на штативе, помещают под нее электрическую плитку или газовую горелку с асбестовой сеткой, включают воду и производят отгонку аммиака при постоянном кипении содержимого колбы. Окончание выделения аммиака устанавливают по исчезновению щелочной реакции отгонной жидкости по красной лакмусовой бумаге. После отгонки наконечник промывают дистиллированной водой для того, чтобы смыть серную кислоту. Приемную колбу снимают и содержимое титруют 0,1 моль/дм3: раствором гидроксида натрия, добавив 3 - 5 капель раствора фенолфталеина или смешанного индикатора. Титрование ведут до перехода бесцветного раствора в слабо-розовый (при использовании фенолфталеина) или из фиолетового в зеленый (при смешанном индикаторе). Параллельно проводят контрольный опыт, оттитровывая 40 см3 0,05 моль/дм3 серной кислоты 0,1 моль/дм3 щелочью.
Обработка результатов. Содержание азота в граммах рассчитывают по формуле
|
|
(53) |
где:
0,0014 - количество азота в граммах, соответствующее 1 см3 0,05 моль/дм3 раствора серной кислоты;
V - количество кубических сантиметров 0,1 моль/дм3 раствора щелочи, пошедшее на титрование 0,05 моль/дм3 раствора серной кислоты в контрольном опыте, см3;
Vl - количество кубических сантиметров 0,1 моль / дм раствора щелочи, пошедшее на титрование 0,05 моль/дм3 раствора свободной серной кислоты в рабочем опыте, см3;
K - поправочный коэффициент к 0,1 моль/дм3 раствору щелочи;
m - масса навески;
Р - масса исследуемого блюда (рациона), г.
Для определения содержания белка в блюде полученное количество азота в граммах умножают на соответствующий коэффициент. В среднем коэффициент пересчета азота на содержание белка в животных пищевых продуктах равен 6,25, в растительных продуктах - 5,5 - 6.
О принятом коэффициенте должно быть указано в рабочем журнале и в бланке результатов анализа.
Пример расчета. В приемную колбу взято 30 см3 точно 0,05 моль/дм3 раствора серной кислоты. На титрование избытка серной кислоты пошло 21,56 см3 точно 0,1 моль/дм3 раствора едкого натра. Навеска второго блюда, взятая для анализа, составляет 0,5 г. Масса блюда 250 г.
Количество азота в блюде в граммах
![]()
Количество белка в блюде: 6,75 × 6,25 = 42,18 г.
2.6.2. Фотометрический метод
Метод основан на минерализации пробы по Къельдалю и фотометрическом измерении интенсивности окраски индофенолового синего, которая пропорциональна количеству аммиака в минерализате.
Аппаратура, материалы, реактивы. Весы лабораторные; спектрофотометр или фотоэлектроколориметр, обеспечивающий измерения в интервалах волн от 315 до 670 нм; мясорубка бытовая или электромясорубка; промывалка стеклянная лабораторная; пробирки стеклянные вместимостью 20 - 25 см3; колбы стеклянные вместимостью 20 - 25 см3; пипетки вместимостью 1 см3 и 5 см3; колбы мерные вместимостью 100, 250, 1000 см3; воронки стеклянные; бюксы; фильтры беззольные, бумажные; кислота серная плотностью 1,84 г/см3; кислота соляная; пероксид водорода; вода дистиллированная; сульфат аммония; известь хлорная; натрия гидроксид; фенол; натрий нитропруссидный; тиосульфат натрия; натрия гипохлорит или натрия дихлоризоцианурат; йодид калия; карбонат натрия безводный.
Подготовка к испытанию. Реактив 1. 10 г фенола и 0,05 нитропруссида натрия растворяют в мерной колбе вместимостью 1000 см3 дистиллированной водой, доводят объем до метки.
Реактив 2. 5 г гидроксида натрия растворяют в дистиллированной воде в мерной колбе вместимостью 1000 см3, после охлаждения добавляют количество исходного раствора гипохлорита натрия из расчета его содержания 0,42 г/дм3 или 0,2 г дихлоризоцианурата натрия и доводят объем колбы дистиллированной водой до метки.
Приготовленные реактивы хранят в темной посуде в холодильнике не более 2 месяцев.
Проведение испытания. Навеску продукта рассчитывают по разности, для этого часть измельченной средней пробы помещают в бюксу, закрывают крышкой и взвешивают с точностью до 0,0002 г. Затем из бюксы скальпелем отбирают 0,4 - 0,5 г продукта на листок беззольного фильтра и вместе с ним осторожно опускают в колбу Къельдаля. Бюксу закрывают, взвешивают и рассчитывают точную массу продукта, взятого для анализа.
Такой же листок беззольного фильтра помещают в контрольную колбу Къельдаля. Затем в обе колбы добавляют 10 см3 концентрированной серной кислоты, 1 - 2 г сульфата калия и проводят минерализацию, для интенсивности процесса периодически добавляя в охлажденную пробу пероксид водорода (5 - 7 см3 в течение всей минерализации). Допускается применение других катализаторов, обеспечивающих точность определения.
После минерализации колбы охлаждают и содержимое количественно переносят в мерные колбы вместимостью 250 см3, после охлаждения объем доводят до метки и содержимое перемешивают.
В мерную колбу вместимостью 100 см3 переносят 5 см3 полученного минерализата и доводят до метки дистиллированной водой. Для проведения цветной реакции 1 см3 разбавленного минерализата вносят в пробирку, затем последовательно добавляют 5 см3 реактива 1 и 5 см3 реактива 2, перемешивают содержимое пробирки. Через 30 мин определяют оптическую плотность растворов на спектрофотометре при длине волны 625 нм или фотоэлектроколориметре.
Контрольный раствор готовят одновременно, используя для этой цели контрольный минерализат.
Стабильность окраски растворов сохраняется в течение одного часа. Температура реактивов при проведении цветной реакции должна быть не ниже 20 °C.
По полученному значению оптической плотности с помощью калибровочного графика находят концентрацию азота.
Обработка результатов. Массовую долю белка (Х) в процентах вычисляют по формуле
|
|
(54) |
где:
С - концентрация азота, найденная по калибровочному графику в соответствии с полученной оптической плотностью, мкг/см3;
m - масса навески пробы, г;
250 - объем минерализата после первого разведения, см3;
5 - объем разбавленного минерализата для вторичного разведения, см3;
100 - объем минерализата после вторичного разведения, см3;
1 - объем раствора, взятый для проведения цветной реакции, см3;
106 - множитель для перевода г в мкг;
100 - множитель для перевода в проценты;
6,25 - коэффициент пересчета на белок.
За окончательный результат принимают среднее арифметическое результатов двух параллельных определений.
Расхождение между параллельными определениями не должно превышать 0,1 % по содержанию азота для мяса и мясопродуктов.
2.7. Определение минеральных веществ (золы)
Метод основан на определении золы, оставшейся после сжигания и прокаливания навески.
Метод предназначен для определения содержания золы при расчете энергетической ценности (калорийности) блюд или рационов.
Аппаратура, материалы. Весы лабораторные; печь муфельная электрическая; эксикатор; фарфоровые тигли с крышкой; треугольник фарфоровый; щипцы тигельные.
Проведение испытания. Навеску блюда (изделия) в количестве 2 - 5 г (в зависимости от предполагаемого содержания в ней воды) берут на аналитических весах с погрешностью не более 0,001 г в предварительно прокаленные в муфельной печи и доведенные до постоянной массы фарфоровые тигли с крышками.
Озоление ведут на газовой горелке при слабом нагреве в закрытом тигле до прекращения выделения газов, следя за тем, чтобы содержимое тигля не выбрасывалось из него. По окончании сухой перегонки тигель приоткрывают и помещают в муфельную печь, нагретую до температуры 600 - 800 °C. Озоление продолжают до тех пор, пока зола не станет серой.
Тигель с золой охлаждают в эксикаторе и взвешивают на аналитических весах. Затем снова прокаливают 20 мин и взвешивают, повторяя эту операцию до тех пор, пока разность между двумя взвешиваниями будет не более 0,0005 г.
Обработка результатов. Содержание золы (Х, %) находят по формуле
|
|
(55) |
где:
m - масса тигля, г;
m1 - масса тигля с остатком после сжигания навески и прокаливания, г;
m2 - масса навески блюда (изделия), г.
2.8. Определение хлористого натрия (поваренной соли)
2.8.1. Аргентометрический метод (метод Мора)
Метод основан на титровании хлоридов в нейтральной среде раствором нитрата серебра в присутствии индикатора хромата калия.
Метод предназначен для определения массовой доли соли в полуфабрикатах, в которых нормируется этот показатель, а также в блюдах (изделиях) в случае разногласий при органолептической оценке.
Аппаратура, материалы, реактивы. Весы лабораторные; мясорубка бытовая; аппарат для встряхивания; термометр ртутный стеклянный со шкалой до 100 °C с ценой деления 1 °C; бюретки вместимостью 10, 25 см3; стаканы химические вместимостью 50, 100, 300, 400 см3; колбы мерные вместимостью 250 см3; колбы конические 100, 250 см3; пипетки вместимостью 10, 25, 50 см3; тигли фарфоровые; воронки стеклянные; бумага фильтровальная; вата гигроскопическая; палочка стеклянная; нитрат серебра, раствор концентрацией 0,05 моль/дм3; хромат калия, раствор с массовой долей 10 % или насыщенный гидроксид натрия или калия, раствор концентрацией 0,1 моль/дм3 (0,1 н); азотная кислота, раствор с массовой долей 10 %; фенолфталеин, спиртовой раствор с массовой долей 1 %; бумага индикаторная универсальная; вода дистиллированная.
Приготовление испытуемого раствора (водной вытяжки). Величину навески (m, г) рассчитывают по формуле
где:
a - заданная массовая доля соли в растворе, который используется для титрования, % (0,2 - 0,5 %);
V - объем мерной колбы, в которую перенесена навеска;
в - предполагаемая массовая доля соли в блюде (изделии), % (для нормально посоленных блюд в зависимости от их вида массовая доля соли составляет от 0,8 до 1,2 %, пересоленных - выше 1,2 % до 3 %).
Массовую долю поваренной соли в блюдах (изделиях) определяют в пробах, подготовленных к анализу, как указано в разделе 4.
При исследовании кулинарных изделий из натурального жареного мяса пробу после удаления костей и сухожилий два раза измельчают в мясорубке с диаметром отверстий решетки 3 - 4,5 мм и тщательно перемешивают. Пробы студней и изделий из рубленого мяса измельчают в мясорубке один раз и тщательно перемешивают.
Из подготовленной пробы блюда (изделия) отбирают навеску и взвешивают с точностью до 0,01 г в химическом стакане или фарфоровой чашке, приливают 40 - 50 см3 горячей (70 - 80 °C) дистиллированной воды, хорошо размешивают продукт стеклянной палочкой и количественно, без потерь, переносят при помощи воронки в мерную колбу вместимостью 200 - 250 см3, смывая прилипшие частицы навески водой.
Колбу доливают дистиллированной водой до половины объема, закрывают пробкой и помещают в аппарат для встряхивания на 15 мин. При отсутствии аппарата для встряхивания содержимое колбы настаивают 25 - 30 мин, периодически взбалтывая.
В связи с тем, что хромат серебра растворяется в кислотах, метод Мора применим лишь в нейтральной или слабощелочной среде, поэтому после охлаждения содержимого до комнатной температуры проверяют реакцию среды по универсальной индикаторной бумаге и при наличии кислот их нейтрализуют 0,1 моль/дм3 (0,1 н) раствором щелочи в присутствии фенолфталеина. Далее колбу доливают дистиллированной водой до метки, закрывают пробкой, содержимое колбы хорошо перемешивают и фильтруют через сухой складчатый фильтр или вату в сухой стакан или колбу. Нейтрализацию водной вытяжки можно провести непосредственно перед титрованием в конической колбе, отобрав аликвотную часть.
Можно также использовать водную вытяжку, приготовленную для определения общей кислотности. В этом случае определенное количество фильтрата, взятое для титрования, предварительно нейтрализуют 0,1 моль/дм3 (0,1 н) раствором щелочи в присутствии фенолфталеина.
Проведение испытания. В зависимости от предполагаемого содержания соли 10 - 50 см3 фильтрата пипеткой переносят в коническую колбу; при наличии кислот фильтрат нейтрализуют 0,1 моль/дм3 (0,1 н) раствором щелочи в присутствии фенолфталеина, приливают 6 - 8 капель 10-процентного или 1 - 2 капли насыщенного раствора хромата калия и титруют 0,05 моль/дм3 (0,05 н) или 0,1 моль/дм3 (0,1 н) раствором нитрата серебра при энергичном взбалтывании до появления в колбе красно-бурого осадка.
При исследовании бульонов 20 см3 бульона титруют 0,05 моль/дм3 (0,05 н) раствором нитрата серебра.
При определении содержания хлорида натрия в кулинарных полуфабрикатах или изделиях из рыбы или в пробах с интенсивной окраской, затрудняющей титрование, навеску рекомендуется озолять. Для этого в тигель берут навеску, подсушивают ее в сушильном шкафу при постепенном повышении температуры до 120 - 140 °C. Высушенную массу осторожно обугливают на газовой горелке до получения остатка темно-серого цвета, легко распадающегося при надавливании стеклянной палочкой. Затем обугленную массу осторожно измельчают палочкой, обрабатывают 4 - 5 раз небольшими порциями горячей воды (80 - 90 °C), каждый раз сливая жидкую часть посредством стеклянной палочки на бумажный фильтр. Фильтрат собирают в мерную колбу вместимостью 250 см3. Остаток в тигле и на фильтре промывают горячей водой до прекращения реакции последних порций фильтрата с нитратом серебра. Для этого небольшую порцию (1 - 2 см3) фильтрата подкисляют в пробирке 1 - 2 каплями раствора азотной кислоты с массовой долей 10 % и прибавляют 1 - 2 капли раствора нитрата серебра. Если фильтрат остается прозрачным, обработку золы горячей водой заканчивают. При необходимости фильтрат в мерной колбе нейтрализуют раствором щелочи в присутствии фенолфталеина, доводят водой до метки, закрывают пробкой и хорошо перемешивают.
Обработка результатов. Массовую долю хлорида натрия (Х) в процентах или в граммах в блюде (изделии) рассчитывают по формуле
|
|
(57) |
где:
V - объем раствора нитрата серебра, израсходованный на титрование, см3;
a - количество хлорида натрия, эквивалентное 1 см3 0,05 моль/дм3 (0,05 н) или 0,1 моль/дм3 (0,1 н) раствора нитрата серебра, г (для 0,05 моль/дм3 - 0,00292, для 0,1 моль/дм3 - 0,00585);
K - поправочный коэффициент к титру раствора нитрата серебра;
Р - масса блюда, г (при определении содержания соли в процентах Р = 100);
V1 - количество фильтрата, взятое для титрования, см3;
m - масса навески, г;
V2 - объем колбы, в которой растворена навеска, см3.
Расхождение между результатами параллельных определений не должно превышать 0,02 %.
За конечный результат принимают среднее арифметическое двух параллельных определений, вычисленное с точностью до 0,1 %.
2.8.2. Электропотенциометрический метод
Метод основан на измерении электропроводности раствора поваренной соли с помощью потенциометра pH-340 или pH-121.
Метод предназначен для определения массовой доли поваренной соли в блюдах (изделиях) в случае разногласий при органолептической оценке.
Аппаратура, материалы, реактивы. Весы лабораторные; аппарат для встряхивания; потенциометр pH-340 или pH-121; мясорубка бытовая; стаканы химические вместимостью 50, 100 и 150 см3; воронки стеклянные диаметром 7 - 9 см; колбы мерные вместимостью 100 см3; палочки стеклянные; пипетка вместимостью 5 см3, градуированная, с ценой деления 0,1 см3; вата гигроскопическая; бумага фильтровальная; хлорид натрия; вода дистиллированная.
Проведение испытания. Перед проведением анализа прибор включают в сеть на 60 мин и настраивают на определение поваренной соли согласно инструкции, прилагаемой к прибору.
Для определения содержания поваренной соли в блюдах и изделиях используют стеклянный электрод марки ЭСЛ-51-Г-04 или ЭСЛ-1Г-05, с помощью которого замеряют концентрацию ионов натрия p№a, и вспомогательный электрод, заполненный насыщенным раствором хлорида калия.
Величину навески для определения содержания поваренной соли в блюдах и изделиях рассчитывают по формуле (56)
где:
a = 0,02 - 0,05 %;
V = 100 см3.
Подготовку пробы к анализу ведут, как указано выше. Нейтрализацию кислот (при их наличии) в данном случае не проводят.
В два химических стакана отбирают около 50 см3 фильтрата*(12). На подвижной столик датчика прибора ставят стаканчик с фильтратом или отстоявшейся жидкостью и надвигают его на электроды датчика так, чтобы кончики их погрузились в жидкость на 15 мм. Перед использованием стеклянный электрод потенциометра выдерживают в течение 8 ч в 0,1 моль/дм3 (0,1 н) раствора хлорида натрия, затем промывают дистиллировальной водой и просушивают фильтровальной бумагой.
________________
*(12) Допускается при подготовке испытуемого раствора сливать отстоявшуюся жидкость из колбы в стаканчики потенциометра без фильтрации.
После минутного выдерживания электродов в испытуемом растворе снимают показания сначала по нижней шкале гальванометра прибора при постановке переключателя на интервал pH от 2 до +14, а затем переключатель ставят на узкий интервал pH и показания снимают по верхней шкале с точностью ±0,1 pH. После каждого замера электроды тщательно промывают дистиллированной водой и просушивают фильтровальной бумагой. Показания шкалы гальванометра прибора переводят с помощью калибровочного графика в величины концентрации поваренной соли.
Калибровочную кривую строят в координатах: на оси абсцисс откладывают концентрацию растворов поваренной соли в процентах, на оси ординат - показания гальванометра прибора - величины pNa.
Для приготовления стандартных растворов 1 г поваренной соли, взвешенной с точностью до 0,001 г, растворяют в 100 см3 дистиллированной воды. Пипеткой отбирают 1,0; 2,0; 4,0 и 5 см3 раствора в мерные колбы вместимостью 100 см3, доводят дистиллированной водой до метки и перемешивают. Замеряют pNa этих растворов. На графике на оси абсцисс откладывают концентрации поваренной соли, равные 0,01; 0,02; 0,04 и 0,5 %, на оси ординат - показания гальванометра прибора.
Для удобства последующих отсчетов калибровочный график наносят на миллиметровую бумагу. Точки пересечения перпендикуляров соединяют. График имеет вид наклонной прямой, идущей слева направо.
Определив значение pNa исследуемого раствора, используя калибровочный график, по двум перпендикулярам - идущему от оси ординат до пересечения с наклонной прямой и по опущенному из точки пересечения с наклонной прямой на ось абсцисс - находят содержание поваренной соли в процентах (или в г на 100 см3), что равноценно содержанию соли во взятой навеске.
Обработка результатов. Массовую долю поваренной соли (Х, %) рассчитывают по формуле
|
|
(58) |
где:
a - количество поваренной соли, найденное по результатам анализа на оси абсцисс, %;
m - навеска продукта, г.
Если рассчитывают содержание соли в порции блюда в граммах, то в числитель формулы вместо 100 подставляют величину Р - массу блюда.
Расхождение между результатами параллельных определений не должно превышать 0,02 %. За конечный результат анализа принимают среднее арифметическое двух параллельных определений, вычисленное с точностью до 0,1 %.
2.9. Определение витамина C (ГОСТ 24556-89)
Метод распространяется на блюда и кулинарные изделия, приготовляемые по "Сборнику рецептур" 1981 г. изд., а также на витаминизированные блюда.
Титриметрический метод с визуальным титрованием используется для определения аскорбиновой кислоты в объектах, дающих светлоокрашенные экстракты, а в объектах, дающих темноокрашенные экстракты, - титриметрический метод с потенциометрическим титрованием.
Титриметрический метод с использованием цистеина служит для определения суммы аскорбиновой и дегидроаскорбиновой кислот (витамина C). Метод применяется при возникновении разногласий в оценке качества.
Методики предназначены для определения витамина C в продуктах с массовой долей не менее 1∙10-3 %.
2.9.1. Титриметрический метод
Метод основан на экстрагировании витамина C раствором кислоты (соляной, метафосфорной или смесью уксусной и метафосфорной) с последующим титрованием визуально или потенциометрически раствором 2,6-дихлорфенолиндофенолята натрия.
Аппаратура, материалы, реактивы. Весы лабораторные; гомогенизатор; pH-метр-милливольтметр лабораторный; мешалка магнитная с плавным регулированием частоты вращения; секундомер с точностью 0,2 с; воронки лабораторные диаметром от 5 до 10 см; колбы мерные лабораторные стеклянные вместимостью 100, 150, 1000, 2000 см3; микробюретка с ценой деления не более 0,01 см3; колбы лабораторные стеклянные вместимостью 50, 100, 250 см3; палочки стеклянные; пипетки мерные лабораторные стеклянные на 1, 2, 5, 10, 20, 25 см3; стаканы лабораторные стеклянные вместимостью 50, 100, 1000 см3; ступка и пестик лабораторные фарфоровые соответственно с наружным диаметром 70 или 90 мм и высотой 90 мм; цилиндры мерные лабораторные стеклянные вместимостью 100, 250 см3; бумага фильтровальная лабораторная; песок кварцевый очищенный и прокаленный; вода дистиллированная; ацетон; 2,6-дихлорфенолиндофенолят натрия, раствор массовой концентрацией 0,250 г/дм3; кислота аскорбиновая должна соответствовать требованиям Государственной фармакопеи СССР, изд. X, растворы массовыми концентрациями 1,0 и 0,1 г/дм3; кислота азотная плотностью 1,14 г/см3, раствор с массовой долей 20 %; кислота метафосфорная, растворы с массовой долей 3 и 6 %. Раствор с массовой долей 3 % готовят в день испытания разбавлением раствора с массовой долей 6 %. Раствор с массовой долей 6 % хранят в холодильнике в течение 10 дней; кислота соляная плотностью 1,19 г/см3, раствор с массовой долей 2 %; кислота уксусная ледяная и раствор с массовой долей 3 %; кислота хлорная, раствор концентрации 0,1 моль/дм3, готовят перед обработкой электродов; кислота этилендиаминтетрауксусная или двунатриевая соль кислоты, раствор с массовой долей 5 %; йодид калия, раствор с массовой долей 1 %, в растворе уксусной кислоты с массовой долей 3 %, готовят перед обработкой электродов; ацетат натрия плавленый, насыщенный раствор (200 г соли растворяют в 300 см3 воды); формальдегид, раствор с массовой долей 36 - 40 %.
Допускается применение импортной аппаратуры, лабораторной посуды и реактивов по классу точности и качеству не ниже отечественных. Для проведения испытания, если нет других указаний, применяют реактивы квалификации "чистый для анализа" и дистиллированную воду или воду эквивалентной чистоты.
Подготовка к испытанию. Приготовление экстрагирующего раствора. В качестве экстрагирующего раствора используют растворы кислот - соляной с массовой долей 2 %, метафосфорной с массовой долей 3 % или смеси уксусной и метафосфорной кислот, которую готовят следующим образом: 15 г метафосфорной кислоты растворяют в 250 см3 дистиллированной воды, прибавляют 40 см3 ледяной уксусной кислоты, доводят водой до объема 500 см3, перемешивают и фильтруют в склянку с притертой пробкой. Хранят в холодильнике не более 10 дней.
Приготовление стандартных растворов аскорбиновой кислоты. Для приготовления раствора аскорбиновой кислоты концентрации 1,0 г/дм3 взвешивают 0,1 г аскорбиновой кислоты с точностью до ±0,0001 г, растворяют в экстрагирующем растворе в мерной колбе вместимостью 100 см3, доводят до метки тем же раствором и перемешивают. Для приготовления раствора концентрации 0,1 г/дм3 вносят пипеткой 10 см3 раствора аскорбиновой кислоты концентрации 1,0 г/дм3 в мерную колбу вместимостью 100 см3, доводят до метки экстрагирующим раствором и перемешивают. Растворы аскорбиновой кислоты неустойчивы, поэтому их готовят перед проведением испытания.
Приготовление раствора 2,6-дихлорфенолиндофенолята натрия и определение его титра. 0,05 г 2,6-дихлорфенолиндофенолята натрия растворяют приблизительно в 150 см3 горячей воды, предварительно прокипяченной в течение 30 мин или содержащей 0,042 г бикарбоната натрия, охлаждают до комнатной температуры, доводят до объема 200 см3 той же охлажденной водой, перемешивают, фильтруют в темную склянку. Раствор хранят в холодильнике не более 10 суток. Титр раствора 2,6-дихлорфенолиндофенолята натрия устанавливают по стандартному раствору аскорбиновой кислоты концентрации 1,0 или 0,1 г/дм3 в день проведения испытания. Для этого в две колбы вместимостью 50 или 100 см3, в которые предварительно прибавлено по 9 см3 воды, вносят пипеткой по 1 см3 раствора аскорбиновой кислоты и быстро титруют раствором 2,6-дихлорфенолиндофенолята натрия до светло-розовой окраски, не исчезающей в течение 15 - 20 с. Одновременно проводят контрольное испытание. Для этого в колбу вместимостью 50 или 100 см3 вносят 1 см3 экстрагирующего раствора, 9 см3 дистиллированной воды и титруют раствором 2,6-дихлорфенолиндофенолята натрия. Титр раствора 2,6-дихлорфенолиндофенолята натрия в граммах аскорбиновой кислоты, эквивалентного одному кубическому сантиметру раствора 2,6-дихлорфенолиндофенолята натрия (Т), вычисляют по формуле
|
|
(59) |
где:
m - масса аскорбиновой кислоты, содержащаяся в 1 см3 стандартного раствора, г;
V1 - объем раствора 2,6-дихлорфенолиндофенолята натрия, израсходованный на титрование стандартного раствора аскорбиновой кислоты, см3;
V2 - объем раствора 2,6-дихлорфенолиндофенолята натрия, израсходованный на контрольное титрование, см3.
Приготовление раствора ацетатного буфера с pH 4. Растворяют 300 г безводного ацетата натрия в 700 см3 дистиллированной воды, добавляют 1000 см3 ледяной уксусной кислоты, перемешивают и с помощью pH-метра устанавливают pH 4, добавляя при необходимости снова кислоту.
Подготовка электродов для потенциометрического титрования. Измерительный платиновый электрод помещают в стакан вместимостью 50 см3 с раствором азотной кислоты, кипятят от 5 до 10 мин, промывают дистиллированной водой и оставляют в воде на 2 - 3 суток. Затем измерительный и вспомогательный электроды опускают в стакан вместимостью 100 см3 с раствором хлорной кислоты и замыкают накоротко (т.е. соединяют клеммы между собой). Через 2 ч электроды вынимают, не размыкая, промывают дистиллированной водой и помещают в стаканы вместимостью 50 см3: измерительный - с раствором йодида калия, вспомогательный - с дистиллированной водой. Через 15 мин электроды размыкают, измерительный электрод промывают дистиллированной водой. Обработке подвергают электроды, не бывшие в употреблении или после перерыва в работе более 6 мес. Хранят в стакане с дистиллированной водой.
Проведение испытаний. Экстрагирование. Для приготовления экстракта навеску пробы массой от 5 до 50 г взвешивают с точностью до 0,01 г.
Величину навески и разбавление определяют из ориентировочного содержания витамина C в продукте, чувствительности метода, а также из того, что проба для титрования должна содержать 0,10 - 0,15 мг аскорбиновой кислоты. Так, при содержании витамина C до 10 мг на 100 г навеска должна составлять 25 - 50 г в объеме 200 - 250 см3, при содержании порядка 40 - 50 мг в 100 г - навеска 5 г в объеме 100 см3. Так, для сиропа из плодов шиповника, плодов шиповника очищенных, пюре из шиповника с сахаром, хвои - 5 г; чая витаминизированного плиточного - 2 - 5 г; хлеба витаминизированного - 60 г; молока витаминизированного - 5 см3; плотной части первого и третьего блюд, плодово-ягодных сладких блюд - 20 - 30 г; жидкой части первого и третьего блюд - 20 - 50 см3; супов-пюре - 20 - 50 г.
Для экстрагирования витамина C из сухих продуктов навеску пробы от 5 до 10 г растирают в ступке с небольшим количеством экстрагирующего раствора кислоты или смеси кислот (не менее 1 см3 раствора на 1 г пробы) и песка, переносят в мерную колбу или мерный цилиндр вместимостью 100 см3, смывая ступку и пестик небольшими порциями экстрагирующего раствора до тех пор, пока объем не достигнет метки. Содержимое выдерживают в течение 10 мин, перемешивают и фильтруют.
Для экстрагирования витамина C из продуктов плотной консистенции навеску пробы от 5 до 50 г гомогенизируют не более 2 мин с небольшим количеством экстрагирующего раствора (не менее 1 см3 раствора на 1 г пробы) и переносят в мерную колбу или цилиндр вместимостью 100 см3, обмывая гомогенизатор небольшими порциями экстрагирующего раствора до тех пор, пока объем не достигнет метки. Содержимое выдерживают в течение 10 мин, перемешивают и фильтруют.
Для экстрагирования витамина C из жидких продуктов навеску пробы от 5 до 50 г переносят в мерную колбу или цилиндр вместимостью 100 см3, обмывая стенки стакана небольшими порциями экстрагирующего раствора до тех пор, пока объем не достигнет метки. Содержимое выдерживают в течение 10 мин, перемешивают и фильтруют.
При исследовании продуктов, содержащих диоксид серы (SO2) (для сульфитированного картофеля) навеску пробы от 5 до 50 г обрабатывают в зависимости от вида продукта, как указано выше, переносят в мерную колбу или цилиндр вместимостью 100 см3, добавляют ацетон в объеме 1/5 части массы навески, перемешивают и доводят объем до метки экстрагирующим раствором.
При исследовании продуктов, фасованных в металлическую тару, навеску пробы от 5 до 30 г обрабатывают, как указано выше, переносят в мерную колбу или цилиндр вместимостью 100 см3 при помощи экстрагирующего раствора, доводят до объема 50 см3 и перемешивают. Через 10 мин прибавляют 10 см3 насыщенного ацетата натрия или 30 см3 раствора этилендиаминтетрауксусной кислоты или ее соли, перемешивают, доводят объем до метки экстрагирующим раствором, снова перемешивают и фильтруют.
Полученные экстракты сразу используют для титрования.
Визуальное титрование. В колбу вместимостью 50 или 100 см3 пипеткой вносят от 1 до 10 см3 экстракта, полученного, как описано выше, доводят объем водой до 10 см3 и титруют раствором 2,6-дихлорфенолиндофенолята натрия до появления слабо-розового окрашивания, не исчезающего в течение 15 - 20 с.
Одновременно проводят контрольное испытание на содержание в продукте редуцирующих веществ. Для этого в колбу помещают такой же объем экстракта, как указано выше, прибавляют равный ему объем ацетатного буферного раствора, раствор формальдегида, равный половине объема буферного раствора, перемешивают и выдерживают в течение 10 мин, закрыв предварительно колбу пробкой. Затем содержимое титруют раствором 2,6-дихлорфенолиндофенолята натрия.
Потенциометрическое титрование. В стакан вместимостью 50 см3 вносят пипеткой объем экстракта, полученного, как указано выше, но не более 25 см3, прибавляют экстрагирующий раствор приблизительно до объема 30 см3 и погружают электроды pH-метра-милливольтметра так, чтобы при перемешивании они не касались магнитного стрежня мешалки. Затем титруют потенциометрически из микробюретки раствором 2,6-дихлорфенолиндофенолята натрия. Раствор 2,6-дихлорфенолиндофенолята натрия прибавляют порциями по 0,1 - 0,2 см3 при постоянном перемешивании. Записывают показания прибора в милливольтах, соответствующие каждому прибавленному раствору 2,6-дихлорфенолиндофенолята натрия. При титровании стрелка прибора сначала отклоняется влево, затем ее движение замедляется, и после точки эквивалентности стрелка отклоняется вправо. Объем раствора 2,6-дихлорфенолиндофенолята натрия, соответствующий точке эквивалентности и, следовательно, израсходованный на титрование объема, устанавливают по максимальной разнице ("скачку") двух соседних показаний прибора или по понтециометрической кривой зависимости величины потенциала в милливольтах от объема раствора 2,6-дихлорфенолиндофенолята натрия в кубических сантиметрах.
Одновременно проводят контрольное титрование на содержание в продукте редуцирующих веществ, как указано выше. Раствор титруют потенциометрически. За результат титрования принимают среднее арифметическое результатов двух титрований одного экстракта. При повторном титровании в области предполагаемой точки эквивалентности раствор 2,6-дихлорфенолиндофенолята натрия прибавляют по 1 - 2 капли.
Обработка результатов. Массовую долю аскорбиновой кислоты (Х, %) вычисляют по формуле
|
|
(60) |
где:
V1 - объем раствора 2,6-дихлорфенолиндофенолята натрия, израсходованный на титрование экстракта, см3;
V2 - объем раствора 2,6-дихлорфенолиндофенолята натрия, израсходованный на контрольное титрование, см3;
Т - титр раствора 2,6-дихлорфенолиндофенолята натрия, г/см3;
V3 - объем экстракта, полученный при экстрагировании витамина C из навески продукта, см3;
V4 - объем экстракта, используемый для титрования, см3;
m - масса навески продукта, г.
За окончательный результат испытания принимают среднее арифметическое результатов двух параллельных определений. Вычисления проводят до четырех значащих цифр после запятой, результат округляют до трех значащих цифр и выражают в виде произведения числа на 10-3.
Расхождение между двумя параллельными определениями не должно превышать 3 % от среднего арифметического значения при доверительной вероятности Р = 0,95.
2.9.2. Титриметрический метод с использованием цистеина
Метод основан на экстрагировании витамина C из продукта раствором метафосфорной кислоты, восстановлении дегидроаскорбиновой кислоты в аскорбиновую кислоту цистеином солянокислым при pH 7,0 - 7,5, устранении влияния редуцирующих веществ в присутствии формальдегида при pH, близком к нулю, и титровании раствором 2,6-дихлорфенолиндофенолята натрия.
Метод применяется при возникновении разногласий в оценке качества сырья и готовой продукции, в том числе витаминизированной, и позволяет определить сумму аскорбиновой и дегидроаскорбиновой кислот.
Аппаратура, материалы, реактивы. Для проведения испытания применяют аппаратуру, материалы и реактивы, как на с. 95, со следующими добавлениями. Термостат электрический с водяной рубашкой или суховоздушный, обеспечивающий поддержание температуры (37 ± 1) °C; колбы мерные лабораторные стеклянные вместимостью 50 см3; калий фосфорнокислый двузамещенный, раствор с массовой долей 45 %; кислота серная, раствор с массовой долей 50 %; альфа-цистеин солянокислый.
Подготовка к испытанию. Приготовление растворов описано выше. Кроме того, используют свежеприготовленный раствор цистеина в растворе соляной кислоты, который готовят следующим образом: 50 мг цистеина растворяют в 4 см3 дистиллированной воды, прибавляют 1 см3 раствора соляной кислоты плотностью 1,19 г/см3 и перемешивают.
Проведение испытания. Экстрагирование. Экстрагирование витамина C из блюд и кулинарных изделий проводят, как указано на с. 97.
От 10 до 20 см3 экстракта пипеткой приливают в мерную колбу вместимостью 50 см3. Одновременно в стакан вместимостью 50 см3 вносят такой же объем экстракта и приливают порциями раствор фосфорнокислого калия двузамещенного до установления pH 7,0 - 7,5, измеряя его с помощью pH-метра. Отмечают объем раствора фосфорнокислого калия. После этого в колбу с экстрактом вносят 50 мг цистеина или его раствор, перемешивают до растворения и прибавляют установленный объем фосфорнокислого калия. Колбу закрывают пробкой и выдерживают в термостате при 37 °C в течение 30 мин. После этого раствор в колбе охлаждают, подкисляя раствором серной кислоты до pH, близкого к нулю, и снова охлаждают. Необходимый для подкисления объем серной кислоты также устанавливают предварительно с помощью pH-метра, используя для этого стакан с экстрактом после прибавления в него фосфорнокислого калия. Раствор в колбе доводят до метки экстрагирующим раствором и перемешивают.
В колбу вместимостью 50 или 100 см3 - для визуального титрования или стакан вместимостью 50 см3 - для потенциометрического титрования вносят пипеткой от 10 до 20 см3 полученного раствора, прибавляют 2 - 3 см3 раствора формальдегида, закрывают крышкой и выдерживают 8 мин, приливают раствор метафосфорной кислоты до объема 30 см3. Затем титруют раствором 2,6-дихлорфенолиндофелята натрия: светлоокрашенные растворы - визуальным титрованием, темноокрашенные - потенциометрическим титрованием, как указано на с. 98, 99.
За результат титрования принимают среднее арифметическое результатов двух титрований одного раствора.
Обработка результатов испытания. Массовую долю витамина C (Х) в процентах вычисляют по формуле
|
|
(61) |
где:
V1 - объем раствора 2,6-дихлорфенолиндофенолята натрия, израсходованный на титрование, см3;
Т - титр раствора 2,6-дихлорфенолиндофенолята натрия, г/см3;
V2 - объем экстракта, полученный при экстрагировании витамина C из навески продукта, см3;
V3 - объем раствора, полученный после восстановления, см3;
V4 - объем экстракта, используемый для восстановления, см3;
V5 - объем раствора, используемый для титрования, см3;
m - масса навески продукта, г.
За окончательный результат испытания принимают среднее арифметическое результатов двух параллельных определений. Вычисление проводят до четырех значащих цифр после запятой, результат округляют до трех значащих цифр и выражают в виде произведения числа на 10-3.
Расхождение между двумя параллельными определениями не должно превышать 3 % от среднего арифметического значения при доверительной вероятности Р = 0,95.
2.10. Определение нитратов и нитритов*(13)
_____________
*(13) Методические указания по определению нитратов и нитритов в продукции растениеводства. М. 1989, № 5048 от 4 июля 1989 г.
Допустимая суточная доза нитратов для человека принимается равной 300 - 325 мг (ср. 312,5 мг).
Допустимые нормы содержания нитратов в продуктах растительного происхождения приведены в табл. 21.
Допустимые уровни содержания нитратов в продуктах растительного происхождения, СаНПиН 42-123-4619-88 от 30 мая 1988 г.*
|
Пищевой продукт |
Содержание нитратов, мк/кг |
|
|
из открытого грунта |
из защищенного грунта |
|
|
Картофель |
250 |
- |
|
Капуста белокочанная |
|
|
|
ранняя (до 1 сентября) |
900 |
- |
|
поздняя |
500 |
- |
|
Морковь |
|
|
|
ранняя (до 1 сентября) |
400 |
- |
|
поздняя |
250 |
- |
|
Томаты |
150 |
300 |
|
Огурцы |
150 |
400 |
|
Свекла столовая |
1400 |
- |
|
Лук репчатый |
80 |
- |
|
Лук-перо |
600 |
800 |
|
Зеленые культуры (салаты, шпинат, щавель, капуста салатная, петрушка, сельдерей, кинза, укроп и т.п.) |
2000 |
3000 |
|
Дыни |
90 |
- |
|
Арбузы |
60 |
- |
|
Перец сладкий |
200 |
400 |
|
Кабачки |
400 |
400 |
|
Тыква (для изготовления консервов для питания детей) |
200 |
- |
|
Виноград столовых сортов |
60 |
- |
|
Яблоки |
60 |
- |
|
Груши |
60 |
- |
_______________
* Дополнения к СанПиН № 4722-88 от 14.11.88.
Для оценки содержания нитратов в свежей плодоовощной продукции и картофеле, поставляемых в открытых автотранспортных средствах, отбор проб для химического анализа производится независимо от формы поставок (навалом, в мешках, в ящиках, в контейнерах и т.п.) непосредственно в транспортном средстве путем взятия 8 выборок по системе двойного конверта из верхнего и более глубоких слоев (например, из нижних ящиков или другой тары при открытии бортов кузова).
Каждая выборка продукции должна иметь массу около 0,5 кг. Если отдельные образцы имеют массу более 0,5 кг (например, кочаны капусты или крупные корнеплоды свеклы), то за выборку принимается отдельный экземпляр (например, кочан капусты).
При поставке продукции в крытом железнодорожном вагоне, автофургоне или другом транспортном средстве, в котором нет свободного доступа к верхнему или другим слоям продукции, или в случае, когда оперативный возврат продукции невозможен, отбор пробы производится при разгрузке транспортного средства путем взятия выборок из разных мест, равномерно распределенных по всему объему партии продукции и удаленных друг от друга на равные расстояния. Независимо от массы партии продукции число выборок равно 12.
При поставке продукции водным транспортом допускается размещение в одном транспортном средстве (барже) нескольких партий при условии их раздельного размещения. В этом случае для химического анализа от каждой партии отбирается отдельная проба. Отбор пробы производится путем взятия 12 выборок из продукции, отобранной от одной партии для проверки ее качества в соответствии с действующим ГОСТ. Если число единиц упаковки (ящики, поддоны, контейнеры) отобранной продукции больше 12, то выборки берутся из произвольно выбранных 12 единиц упаковки. Если число единиц упаковки меньше 12, то из каждой единицы упаковки отбирается по несколько выборок. Места выборок из одной упаковки (например, из контейнера) должны быть равномерно рассредоточены по ее объему.
В случае возникновения разногласий между получателем и поставщиком отбор проб продукции проводят в соответствии с ГОСТ 1724-85, ГОСТ 1721-85, ГОСТ 26768-85, ГОСТ 26767-85, ГОСТ 1722-85, ГОСТ 6766-85, ГОСТ 7194-81, ГОСТ 26545-85, ГОСТ 1725-85, ГОСТ 1726-85, ГОСТ 1723-67, ГОСТ 7177-80, ГОСТ 7178-85, ГОСТ 7975-68, ГОСТ 13907-86, ГОСТ 13908-68, ГОСТ 7967-68, ГОСТ 7968-68, ГОСТ 7977-67, с последующим проведением анализа только стандартной части экспертируемой продукции. Пробы от стандартной части продукции отбирают методом конверта (по 0,5 кг из каждой точки отбора).
Пробы готовят к анализу следующим образом:
Картофель. Клубни моют водой, вытирают чистой тканью досуха и разрезают крестообразно вдоль оси "столон - ростовая часть" на 4 равные части. От каждого клубня берут четвертую часть, отобранный материал используют для анализа.
Корнеплоды. Корнеплоды моют водой, вытирают чистой тканью досуха, срезают шейку и тонкий конец корня и разрезают крестообразно вдоль вертикальной оси на 4 равные части. Доли, представляющие четвертую часть от каждого корнеплода, используют для анализа.
Капуста. Кочаны разрезают крестообразно вдоль вертикальной оси на 4 или 8 равных частей и берут соответственно по 1/4 или 1/8 части от каждого кочана в пробу для анализа. При этом отбрасывают верхние несъедобные листья и остаток кочерыги.
Луковичные растения. Отбрасывают несъедобные части. С луковиц удаляют чешуи, срезают и отбрасывают основания корня и сухую шейку, разрезают их крестообразно вдоль вертикальной оси на 4 равные части и от каждой луковицы четвертую часть берут в пробу для анализа.
Томаты, огурцы, кабачки. Плоды моют водой, вытирают чистой тканью досуха, удаляют плодоножки и разрезают крестообразно вдоль оси на 4 равные части. От каждого плода в пробу для анализа берут по 1/4 части.
Бахчевые культуры. Плоды разрезают вдоль оси на сегменты шириной 6 - 8 см по окружности плода и в пробу для анализа от каждого плода берут по 2 - 4 сегмента с противоположных сторон таким образом, чтобы в их число попали и затемненные, и освещенные солнцем части. С отобранных частей плода снимают верхний слой, не употребляемый в пищу, удаляют семена.
Перец сладкий. Плоды моют водой, вытирают чистой тканью досуха, разрезают крестообразно вдоль оси на 4 равные части и берут в пробу для анализа по 1/4 части от каждого плода. При этом вырезают и отбрасывают семена и остаток плодоножки.
Зеленые овощи (салат, шпинат, капуста салатная, петрушка, щавель, сельдерей, кинза, укроп и т.д.). Обрезают и отбрасывают несъедобные части растений. Растения моют водой и подсушивают сначала между листами фильтровальной бумаги или слоями чистой ткани, а затем на воздухе.
Яблоки, груши. Плоды моют водой, вытирают чистой тканью досуха, разрезают крестообразно вдоль оси на 4 равные части и берут в пробу для анализа по 1/4 части от каждого плода. При этом вырезают и отбрасывают остаток семенного гнезда и плодоножку.
Виноград. Ягоды винограда отделяют от веток, моют водой и сушат на листе фильтровальной бумаги.
Примечание: пробы необходимо готовить в количестве не менее двух, так как в случае повышенного содержания нитратов может возникнуть необходимость повторения анализа*(14).
______________
*(14) "Временные методические указания по отбору проб растительной продукции и кормов для определения нитратов и остаточных количеств пестицидов", утвержденные 3 апреля 1989 г.
Порядок отбора и подготовки проб полуфабрикатов, готовых блюд и кулинарных изделий изложен в прил. 1, 2.
2.10.1. Ионометрический метод определения нитратов
Метод основан на извлечении нитратов из анализируемого материала раствором алюмокалиевых квасцов с последующим измерением их концентрации в полученной вытяжке с помощью ионоселективного электрода. Для ускорения анализа вместо вытяжки можно использовать сок анализируемой продукции, разбавленный раствором алюмокалиевых квасцов. При анализе капусты для разрушения примесей, мешающих определению нитратов, проводят их окисление марганцевокислым калием.
Метод используют при количественном определении нитратов в овощах, плодах и ягодах.
Метод непригоден, если содержание хлоридов в анализируемом материале более чем в 25 раз превышает содержание нитратов при их концентрации до <...> мг/кг и в 50 раз - при более высоких.
Нижний предел обнаружения нитратов - 6 мг на 1 дм3 анализируемого раствора. Предел надежного определения нитратов в анализируемой пробе - 30 млн-1 (мг/кг).
Аппаратура, материалы, реактивы. Иономер типа И-120М, И-115М, ЭВ-74; милливольтметры pH-340, pH-121; нитратомер НМ-002 или аналогичный прибор с погрешностью измерения не более 5 мВ (0,05 PNO2); ионоселективный нитратный электрод ЭМ-NO-3 - 0,1 , ЭИМ-11 или другой электрод, имеющий такие же метрологические характеристики; электрод вспомогательный лабораторный хлорсеребряный ЭВЛ-1М1 или ЭВЛ-1МЗ; весы лабораторные; колбы мерные вместимостью 50, 100 и 1000 см3; мерный цилиндр вместимостью 50 см3; пипетки вместимостью 1, 5, 10, 50 и 100 см3; стаканы лабораторные вместимостью 100, 200, 500 и 1000 см3; ступка фарфоровая с пестиком; капельница; встряхиватель ВВ-1 или блок экстрагирования БЭ-1; мешалка лабораторная электромеханическая или магнитная; мезгообразователь МЛ-1; пластмассовая терка; механическая или электромеханическая мясорубка; ножницы; нож; гомогенизатор с частотой вращения ножевой системы не менее 6000 мин-1; измельчитель РТ-1 и РТ-2; соковыжималка с электрическим удалением выжимок; механическая соковыжималка; шпатель; стеклянные палочки; квасцы алюмокалиевые (KAl(SO4)2∙12H2O); хлорид калия; нитрат калия; перманганат калия; кислота борная; вода дистиллированная; пероксид водорода, раствор с массовой долей 35 %; бумага масштабно-координатная марки Д2.
Подготовка к испытанию. 1. Раствор алюмокалиевых квасцов с массовой долей 1 % (экстрагирующий раствор): 10 г алюмокалиевых квасцов, взвешенных с точностью до 0,1 г, растворяют дистиллированной водой в мерной колбе вместимостью 1000 см3 и доводят объем до метки. Раствор хранят в склянке с притертой пробкой не более года. При появлении мути или осадка его заменяют на свежеприготовленный.
2. Экстрагирующий раствор для определения нитратов в капусте: 10 г алюмокалиевых квасцов, взвешенных с точностью до 0,1 г, растворяют дистиллированной водой в мерной колбе вместимостью 1000 см3, туда же добавляют 1 г перманганата калия, взвешенного с точностью до 0,01 г, и 0,6 см3 концентрированной серной кислоты. Полученную смесь взбалтывают до растворения всех ингредиентов, раствор доводят до метки дистиллированной водой и хранят в склянке с притертой пробкой не более одного года.
3. Приготовление основного раствора нитрата калия
концентрации С(KNO3) = 0,1
моль/дм3 (![]() = -lgCNO3
= 1): 10,11 г нитрата калия, высушенного при температуре 110 - 120 °C до
постоянной массы и взвешенного с точностью до 0,001 г, растворяют
экстрагирующим раствором (п. 1) в мерной колбе 1000 см3 и доводят
объем до метки тем же раствором. Раствор хранят в склянке с притертой пробкой
не более года, при появлении мути или осадка его заменяют на
свежеприготовленный.
= -lgCNO3
= 1): 10,11 г нитрата калия, высушенного при температуре 110 - 120 °C до
постоянной массы и взвешенного с точностью до 0,001 г, растворяют
экстрагирующим раствором (п. 1) в мерной колбе 1000 см3 и доводят
объем до метки тем же раствором. Раствор хранят в склянке с притертой пробкой
не более года, при появлении мути или осадка его заменяют на
свежеприготовленный.
4. Приготовление растворов сравнения нитрата калия: растворы готовят из основного раствора нитрата калия (п. 3). В день проведения анализа, используя для разбавления раствор алюмокалиевых квасцов (п. 1), который применяется для анализа. Растворы сравнения используют для градуировки прибора и построения градуировочного графика.
4.1. Раствор сравнения с концентрацией С(NO-3) = 0,01 моль/дм3 (![]() = 2): основной раствор нитрата калия
разбавляют в 10 раз раствором алюмокалиевых квасцов. Для этого в мерную колбу
вместимостью 100 см3 отбирают пипеткой 10 см3 основного
раствора с
= 2): основной раствор нитрата калия
разбавляют в 10 раз раствором алюмокалиевых квасцов. Для этого в мерную колбу
вместимостью 100 см3 отбирают пипеткой 10 см3 основного
раствора с ![]() = 1, доводят до метки раствором
алюмокалиевых квасцов и перемешивают.
= 1, доводят до метки раствором
алюмокалиевых квасцов и перемешивают.
4.2. Раствор сравнения с концентрацией С(NO-3) = 0,001 моль/дм3 (![]() = 3): раствор, приготовленный по п.
4.1, разбавляют в 10 раз раствором алюмокалиевых квасцов, как указано в п. 4.1.
= 3): раствор, приготовленный по п.
4.1, разбавляют в 10 раз раствором алюмокалиевых квасцов, как указано в п. 4.1.
4.3. Раствор сравнения с концентрацией С(NO-3) = 0,0001 моль/дм3
(![]() = 4): раствор, приготовленный по п.
4.2, разбавляют в 10 раз раствором алюмокалиевых квасцов.
= 4): раствор, приготовленный по п.
4.2, разбавляют в 10 раз раствором алюмокалиевых квасцов.
5. Мембранный ионоселективный нитратный электрод и
хлорсеребряный электрод сравнения готовят к работе в соответствии с
инструкциями, прилагаемыми к ним. В промежутках между исследованиями мембранный
ионоселективный электрод погружают в раствор с ![]() = 4. Если перерывы в работе составляют
сутки и более, его хранят в растворе нитрата калия с концентрацией С(NO-3) = 0,001 моль/дм3.
При длительных перерывах между исследованиями (более пяти суток) электрод
хранят на воздухе и перед началом работы вымачивают 1 - 2 часа в растворе
нитрата калия с концентрацией С(NO-3)
= 0,1 моль/дм3. в обоих случаях перед началом измерений электрод
промывают в дистиллированной воде не менее 3 раз. Хлорсеребряный электрод
сравнения в перерывах между исследованиями погружают в стакан с
дистиллированной водой.
= 4. Если перерывы в работе составляют
сутки и более, его хранят в растворе нитрата калия с концентрацией С(NO-3) = 0,001 моль/дм3.
При длительных перерывах между исследованиями (более пяти суток) электрод
хранят на воздухе и перед началом работы вымачивают 1 - 2 часа в растворе
нитрата калия с концентрацией С(NO-3)
= 0,1 моль/дм3. в обоих случаях перед началом измерений электрод
промывают в дистиллированной воде не менее 3 раз. Хлорсеребряный электрод
сравнения в перерывах между исследованиями погружают в стакан с
дистиллированной водой.
Проведение испытания. Пробы для анализа измельчают с помощью терки механической, электромеханической мясорубки или мезгообразователя. Зеленые культуры режут ножницами или ножом до частиц размером 0,5 - 1,0 см или измельчают на мясорубке. 10,0 г измельченного материала взвешивают с точностью до 0,01 г, помещают в стакан гомогенизатора или измельчителя, приливают 50 см3 раствора алюмокалиевых квасцов и гомогенизируют в течение 1 мин при частоте вращения 6000 мин-1.
При отсутствии гомогенизатора или измельчителя навеску продукта помещают в стакан вместимостью 100 - 200 см3, приливают 50 см3 раствора алюмокалиевых квасцов и перемешивают с помощью мешалки в течение 3 мин.
При анализе материала, содержащего твердые ткани, и отсутствии гомогенизатора навеску массой 10,0 г растирают в ступке с прокаленным песком или битым стеклом до однородной массы, переносят ее с помощью 50 см3 раствора алюмокалиевых квасцов в стакан вместимостью 100 - 200 см3, перемешивают в течение трех минут с помощью мешалки. Вместо растирания в ступке с песком или битым стеклом возможно 15-минутное нагревание суспензии на кипящей водяной бане с последующим охлаждением и доведением до метки.
В суспензии, приготовленной одним из описанных способов, измеряют концентрацию нитрат-ионов.
При анализе капусты 10,0 г измельченного материала взвешивают с точностью до 0,1 г, помещают в стакан вместимостью 100 см3, наливают 50 см3 раствора алюмокалиевых квасцов (п. 2), перемешивают с помощью мешалки в течение 3 мин. Затем при перемешивании добавляют по каплям (2 - 3 капли) раствора пероксида водорода до обесцвечивания раствора. В полученной суспензии измеряют концентрацию нитрат-ионов.
Для овощей, плодов и ягод, кроме зеленых культур, с целью ускорения и снижения трудоемкости анализа можно использовать сок. Для получения сока пробы пропускают через электромеханическую соковыжималку. Сок собирают в мерную емкость и перемешивают.
При анализе всех культур, кроме капусты, от полученного сока с помощью пипетки отбирают аликвотную часть объемом 10 см3, измеренную с точностью до 0,1 см3, помещают ее в стакан вместимостью 100 - 200 см3, прибавляют 50 см3 раствора алюмокалиевых квасцов, перемешивают и в полученном растворе измеряют концентрацию нитрат-ионов.
При анализе капусты в стакан вместимостью 100 - 200 см3 помещают 10 см3 сока, измеренного с точностью до 0,1 см3, прибавляют 50 см3 раствора алюмокалиевых квасцов (п. 2) и перемешивают в течение 3 мин. Затем при перемешивании добавляют по каплям (2 - 3 капли) раствор пероксида водорода до обесцвечивания раствора, после чего измеряют концентрацию нитрат-ионов с помощью электрода ЭМ-NO-3-01.
Градуировка иономера при измерении концентрации нитратов в
единицах ![]() . После подключения прибора к
электросети при работе с электродом ЭМ-NO-3-1
нажимают клавишу 1 + 4.
. После подключения прибора к
электросети при работе с электродом ЭМ-NO-3-1
нажимают клавишу 1 + 4.
Подготовленные к работе нитратный и хлорсеребряный электроды
ополаскивают дистиллировальной водой, промокают фильтровальной бумагой и
погружают в раствор сравнения с концентрацией С(NO-3)
= 0,0001 моль/дм3 (![]() = 4).
Переключатель рода термокомпенсации иономера должен находиться при этом в
положении "Ручн", а ручки "Температура раствора" и
"pX" повернуты в крайнее левое положение. При замере
= 4).
Переключатель рода термокомпенсации иономера должен находиться при этом в
положении "Ручн", а ручки "Температура раствора" и
"pX" повернуты в крайнее левое положение. При замере ![]() нажимают клавишу "pX", при
отключении цепи - клавишу "t". Ручкой "Калибровка"
устанавливают стрелку прибора на значение, равное 4 (по средней шкале прибора).
Затем убирают раствор с концентрацией С(NO-3)
= 0,0001 моль/дм3, ополаскивают электроды дистиллировальной водой,
промокают их фильтровальной бумагой, чтобы убрать капли воды, и погружают
электроды в раствор с концентрацией С(NO-3)
= 0,01 моль/дм3 (
нажимают клавишу "pX", при
отключении цепи - клавишу "t". Ручкой "Калибровка"
устанавливают стрелку прибора на значение, равное 4 (по средней шкале прибора).
Затем убирают раствор с концентрацией С(NO-3)
= 0,0001 моль/дм3, ополаскивают электроды дистиллировальной водой,
промокают их фильтровальной бумагой, чтобы убрать капли воды, и погружают
электроды в раствор с концентрацией С(NO-3)
= 0,01 моль/дм3 (![]() = 2)
= 2)
С помощью ручки "Крутизна" устанавливают стрелку
прибора на значение, равное двум (средней шкалы прибора). Если с помощью ручки
"Крутизна" не удается вывести на значение, равное двум, то используют
ручку "Температура раствора". Затем повторяют настройку прибора по раствору
![]() = 4 и
= 4 и ![]() = 2 до тех
пор, пока стрелка прибора не будет показывать требуемые значения. Перед каждой
проверкой настройки иономера электроды выдерживают в растворе с концентрацией С(NO-3) = 0,0001 моль/дм3
3 - 4 мин и каждый раз измерения проводят в свежей порции раствора сравнения.
При переносе из концентрированного в разбавленный раствор электроды
ополаскивают дистиллировальной водой и сушат фильтровальной бумагой. Проверку
градуировки прибора проверяют по раствору
= 2 до тех
пор, пока стрелка прибора не будет показывать требуемые значения. Перед каждой
проверкой настройки иономера электроды выдерживают в растворе с концентрацией С(NO-3) = 0,0001 моль/дм3
3 - 4 мин и каждый раз измерения проводят в свежей порции раствора сравнения.
При переносе из концентрированного в разбавленный раствор электроды
ополаскивают дистиллировальной водой и сушат фильтровальной бумагой. Проверку
градуировки прибора проверяют по раствору ![]() = 3. Отклонение
от контрольного значения не должно превышать ±0,04
= 3. Отклонение
от контрольного значения не должно превышать ±0,04![]() .
.
Закончив градуировку прибора, электроды погружают в
испытуемый раствор и снимают показания в единицах ![]() . Показания прибора считывают не ранее
чем через 1 мин после прекращения дрейфа показаний прибора. Температура
испытуемых проб и растворов сравнения должна быть одинаковой. Настройку прибора
проверяют не менее трех раз в течение рабочего дня, используя каждый раз свежие
порции растворов сравнения.
. Показания прибора считывают не ранее
чем через 1 мин после прекращения дрейфа показаний прибора. Температура
испытуемых проб и растворов сравнения должна быть одинаковой. Настройку прибора
проверяют не менее трех раз в течение рабочего дня, используя каждый раз свежие
порции растворов сравнения.
Полученные значения ![]() переводят в мг/кг NO-3
по формуле (62) или по табл. 22 - 27.
переводят в мг/кг NO-3
по формуле (62) или по табл. 22 - 27.
Градуировку прибора типа pH-метр-милливольтметр (pH-340) проводят следующим образом. Перед настройкой прибора следует поставить тумблер "Род работ" в положение "pH", измерительный нитратный электрод к гнезду "Всп", а вспомогательный хлорсеребряный электрод подключить к гнезду "Изм", т.к. измеряется концентрация аниона (NO-3), а прибор рассчитан на измерение водородного иона.
Предел измерения для электрода ЭМ-NO-3-01 2 + 5 pH. Работа ведется при размахе 3 pH, или 300 мВ. На стекле шкалы делают перецифровку - соответственно цифрам на верхней шкале прибора 3, 2, 1 пишут слева направо 4, 3, 2, 1.
Подготовленные к работе электроды ополаскивают
дистиллированной водой, промокают фильтровальной бумагой и помещают в раствор
сравнения с концентрацией С(NO-3)
= 0,0001 моль/дм3 (![]() = 4) и с
помощью ручек "Еи-грубо" на задней стенке прибора и
"Еи-точно" на верхней панели прибора устанавливают стрелку прибора на
верхней шкале на значение 4 (по измененному обозначению). Затем ополаскивают
электроды дистиллированной водой, промокают фильтровальной бумагой и погружают
электрод в раствор сравнения с концентрацией С(NO-3)
= 0,01 моль/дм3 (
= 4) и с
помощью ручек "Еи-грубо" на задней стенке прибора и
"Еи-точно" на верхней панели прибора устанавливают стрелку прибора на
верхней шкале на значение 4 (по измененному обозначению). Затем ополаскивают
электроды дистиллированной водой, промокают фильтровальной бумагой и погружают
электрод в раствор сравнения с концентрацией С(NO-3)
= 0,01 моль/дм3 (![]() = 2), с
помощью ручки "S" устанавливают стрелку прибора на значение, равное
двум (по измененному обозначению); если с помощью ручки "S" это
сделать невозможно, то используют тумблер "Температура раствора".
Затем повторяют настройку прибора по растворам сравнения с
= 2), с
помощью ручки "S" устанавливают стрелку прибора на значение, равное
двум (по измененному обозначению); если с помощью ручки "S" это
сделать невозможно, то используют тумблер "Температура раствора".
Затем повторяют настройку прибора по растворам сравнения с ![]() = 4 и
= 4 и ![]() = 2 до тех
пор, пока стрелка прибора не будет показывать требуемые значения. Перед
проверкой настройки иономера электроды необходимо выдержать в растворе с
концентрацией С(NO-3)
= 0,0001 моль/дм3 3 - 4 мин и каждый раз проводить измерение в
свежей порции растворов сравнения.
= 2 до тех
пор, пока стрелка прибора не будет показывать требуемые значения. Перед
проверкой настройки иономера электроды необходимо выдержать в растворе с
концентрацией С(NO-3)
= 0,0001 моль/дм3 3 - 4 мин и каждый раз проводить измерение в
свежей порции растворов сравнения.
При переносе из концентрированного раствора в разбавленный
электроды ополаскивают дистиллированной водой и сушат фильтровальной бумагой.
Проверку градуировки прибора проводят по раствору с ![]() = 3. Показания
прибора считывают не ранее чем через 1 мин после прекращения дрейфа показаний
прибора. Температура испытуемых проб и растворов сравнения должна быть
одинаковой. Настройку прибора проверяют не менее трех раз в течение рабочего
дня, используя каждый раз свежие порции растворов сравнения. Отклонения от
контрольного значения не должны превышать ±0,04
= 3. Показания
прибора считывают не ранее чем через 1 мин после прекращения дрейфа показаний
прибора. Температура испытуемых проб и растворов сравнения должна быть
одинаковой. Настройку прибора проверяют не менее трех раз в течение рабочего
дня, используя каждый раз свежие порции растворов сравнения. Отклонения от
контрольного значения не должны превышать ±0,04![]() .
.
Закончив градуировку прибора, электроды погружают в испытуемый
раствор и снимают показания в ![]() . В течение рабочего дня градуировку
прибора периодически повторяют по растворам сравнения. Полученные при анализах
значения
. В течение рабочего дня градуировку
прибора периодически повторяют по растворам сравнения. Полученные при анализах
значения ![]() переводят в мг/кг NO-3
по формуле (62) или по табл. 22 - 27.
переводят в мг/кг NO-3
по формуле (62) или по табл. 22 - 27.
Измерение концентрации иона нитрата в режиме "мВ". Определение проводят, используя калибровочный график и снимая показания ЭДС в "мВ". В этом случае нитратный электрод (для любых милливольтметров) подключают к гнезду "Изм", а хлорсеребряный электрод - к гнезду "Всп". Тумблер "Род работ" ставят в положение "мВ" и ЭДС электродной пары измеряют в растворах сравнения и анализируемых.
Перед началом работы измеряют показания растворов сравнения в порядке возрастания концентрации, начиная с меньшей, с концентрации С(NO-3) = 0,0001 моль/дм3.
Перед погружением электродов исследуемые пробы взбалтывают. Показания прибора считывают не ранее чем через 1 мин после прекращения дрейфа показаний прибора. Температура испытуемых проб и растворов сравнения должна быть одинаковой. Приборы настраивают не менее трех раз в течение рабочего дня, используя каждый раз свежие порции растворов сравнения.
После каждого измерения электроды ополаскивают дистиллированной водой и промокают фильтровальной бумагой.
Содержание нитратов в пробах находят, используя
калибровочный график, построенный на миллиметровой бумаге. По оси абсцисс
откладывают величины ![]() , соответствующие растворам сравнения
нитрата калия в молях:
, соответствующие растворам сравнения
нитрата калия в молях:
с концентрацией С(NO-3)
= 0,1 моль/дм3 (![]() = 1)
= 1)
с концентрацией С(NO-3)
= 0,01 моль/дм3 (![]() = 2)
= 2)
с концентрацией С(NO-3)
= 0,001 моль/дм3 (![]() = 3)
= 3)
с концентрацией С(NO-3)
= 0,0001 моль/дм3 (![]() = 4)
= 4)
по оси ординат - ЭДС, мВ. По калибровочному графику находят
значения величины с концентрацией ![]() и с помощью уравнения (62) или табл. 22
- 27 определяют содержание нитрат-ионов
(мк/кг) в исследуемом объекте.
и с помощью уравнения (62) или табл. 22
- 27 определяют содержание нитрат-ионов
(мк/кг) в исследуемом объекте.
При работе с приборами, имеющими преобразователи величины ![]() или моль/дм3 в значении концентрации
иона нитрата в исследуемой продукции, настройку проводят непосредственно в
единицах массовой доли нитрата в миллионных долях (мг/кг) по растворам
сравнения с концентрацией С(NO-3)
= 0,0001 моль/дм3 (
или моль/дм3 в значении концентрации
иона нитрата в исследуемой продукции, настройку проводят непосредственно в
единицах массовой доли нитрата в миллионных долях (мг/кг) по растворам
сравнения с концентрацией С(NO-3)
= 0,0001 моль/дм3 (![]() = 4) и С(NO-3) = 0,01 моль/дм3 (
= 4) и С(NO-3) = 0,01 моль/дм3 (![]() = 2) используя
раствор С(NO-3) =
0,001 моль/дм3 для контроля. При настройке таких приборов значение
массовой доли нитратов берется из вспомогательных табл. 22 - 27 или
вычисляется по формуле (62).
= 2) используя
раствор С(NO-3) =
0,001 моль/дм3 для контроля. При настройке таких приборов значение
массовой доли нитратов берется из вспомогательных табл. 22 - 27 или
вычисляется по формуле (62).
При работе с иономером НМ-002, который не может
преобразовывать величины более 1900 млн.-1 (мг/кг), при его
настройке следует загрублять показания в 10 раз. Так, например, при настройке
прибора по табл. 22, согласно которой
раствору сравнения с ![]() = 4
соответствует массовая доля нитрат-ионов 36 млн.-1 (мг/кг), а
раствору с
= 4
соответствует массовая доля нитрат-ионов 36 млн.-1 (мг/кг), а
раствору с ![]() = 2 - 3596
млн.-1 (мг/кг), с помощью кнопок "К1" и "К2"
прибор настраивают на значения 3,6 и 359 млн.-1 (мг/кг)
соответственно. Контроль производят по раствору с
= 2 - 3596
млн.-1 (мг/кг), с помощью кнопок "К1" и "К2"
прибор настраивают на значения 3,6 и 359 млн.-1 (мг/кг)
соответственно. Контроль производят по раствору с ![]() = 3 (36 млн.-1).
Таким же образом настраивают прибор и по другим таблицам. При определении
содержания нитратов в исследуемых культурах результат увеличивают в 10 раз.
= 3 (36 млн.-1).
Таким же образом настраивают прибор и по другим таблицам. При определении
содержания нитратов в исследуемых культурах результат увеличивают в 10 раз.
Измерения концентрации нитрат-ионов с помощью электрода ЭИМ-11 и иономера ЭВ-74 проводят в соответствии с техническим описанием и инструкцией по эксплуатации электрода ЭИМ-11.
Прежде чем приступить к измерению, необходимо: вымочить электрод ЭИМ-11 в течение 1 - 2 суток в растворе нитрата калия с концентрацией С(NO-3) = 0,1 моль/дм3, промыть электроды дистиллированной водой и удалить с них капли воды фильтровальной бумагой; включить иономер ЭВ-74 в сеть (при этом в правом верхнем углу панели прибора загорится индикатор) и подсоединить электрод ЭИМ-11 к гнезду "Изм", а вспомогательный электрод - к гнезду "Всп"; нажать клавиши "t", "анион-катион" и "9-14", ручку "Калибровка" вывести в крайнее левое положение, для чего вращают ее против часовой стрелки до упора.
Градуировку иономера проводят по двум растворам сравнения с ![]() = 2 и
= 2 и ![]() = 4. Начать
градуировку следует с раствора
= 4. Начать
градуировку следует с раствора ![]() = 4. С этой
целью электрод ЭИМ-11 и вспомогательный электрод погружают в раствор сравнения
с концентрацией С(NO-3)
= 0,0001 моль/дм3 (
= 4. С этой
целью электрод ЭИМ-11 и вспомогательный электрод погружают в раствор сравнения
с концентрацией С(NO-3)
= 0,0001 моль/дм3 (![]() = 4) и, нажав
клавишу "pX" ручкой "калибровка" устанавливают стрелку
прибора на любое оцифрованное деление в центре шкалы (например, 2). Присваивают
этому делению величину 4,00
= 4) и, нажав
клавишу "pX" ручкой "калибровка" устанавливают стрелку
прибора на любое оцифрованное деление в центре шкалы (например, 2). Присваивают
этому делению величину 4,00 ![]() . Нажимают клавишу "t". Затем
вынимают электроды из раствора, промокают фильтрованной бумагой и погружают в
раствор с
. Нажимают клавишу "t". Затем
вынимают электроды из раствора, промокают фильтрованной бумагой и погружают в
раствор с ![]() = 2. Нажимают
клавишу "pX". Стрелка прибора должна отклониться вправо примерно на
два больших деления. Ручкой "крутизна" и "температура"
устанавливают стрелку прибора на отметку 4, присвоив ей величину 2,00
= 2. Нажимают
клавишу "pX". Стрелка прибора должна отклониться вправо примерно на
два больших деления. Ручкой "крутизна" и "температура"
устанавливают стрелку прибора на отметку 4, присвоив ей величину 2,00 ![]() . Нажимают клавишу "t".
Вынимают электроды из раствора, промывают их дистиллированной водой и промокают
фильтровальной бумагой.
. Нажимают клавишу "t".
Вынимают электроды из раствора, промывают их дистиллированной водой и промокают
фильтровальной бумагой.
Все описанные операции повторяют до тех пор, пока стрелка не будет точно устанавливаться на отметках 2,00 и 4,00 вновь присвоенной шкалы прибора.
Проверку калибровки проводят, погружая электроды в раствор
сравнения с ![]() = 3,00.
Стрелка прибора должна останавливаться на делении 3,00. Допустимые отклонения
±0,04
= 3,00.
Стрелка прибора должна останавливаться на делении 3,00. Допустимые отклонения
±0,04 ![]() . Проверку калибровки проводят не менее
3 раз в течение рабочего дня.
. Проверку калибровки проводят не менее
3 раз в течение рабочего дня.
Для измерения концентрации нитратов в испытуемом растворе электроды погружают в раствор и нажимают клавишу "pX". Показания прибора считывают не ранее чем через 1 мин после прекращения дрейфа показаний прибора. Перед каждым измерением электроды ополаскивают водой и промокают фильтровальной бумагой.
Электрод ЭИМ-11 работает в отрицательной области "мВ" (700 - 900 "мВ"). При работе с этим электродом в режиме "мВ" следует нажимать клавиши "Анионы/катионы" и диапазон 4 - 9. Построение градуировочного графика и измерения проводят так же, как и при работе с электродом ЭМ-NO-3-01.
Полученные значения ![]() переводят в мг/кг NO-3
по формуле (62) или по табл. 22 - 27.
переводят в мг/кг NO-3
по формуле (62) или по табл. 22 - 27.
Обработка результатов испытания. Если при анализе использовалась навеска измельченной пробы, то массовую долю нитратов в испытуемом материале (Х) в миллионных долях (млн.-1, мг/кг) рассчитывают по формуле
где:
62 - молярная масса иона нитрата, г;
m - масса пробы, взятой для анализа, г;
V - объем экстрагирующего раствора, см3;
![]() - концентрация нитрата в вытяжке,
моль/дм3;
- концентрация нитрата в вытяжке,
моль/дм3;
1000 - коэффициент перевода дм3 в см3;
W - массовая доля воды в пробе, %;
100 - коэффициент перевода % в доли единиц;
I - плотность воды, г/см3;
106 - коэффициент перевода долей единицы в миллионные доли (млн.-1, мг/кг).
Принимая V = 50 см3, m = 10 г и проведя соответствующие сокращения и преобразования, получаем следующую формулу для расчета
|
|
(63) |
При разбавлении вытяжки результат анализа увеличивают во столько раз, во сколько раз была разбавлена вытяжка.
Расчеты по приведенным уравнениям можно исключить, используя
табл. 22 - 23 для перевода величин ![]() в массовую долю нитрата в
анализируемой пробе. Данные таблицы составлены с учетом среднего содержания
влаги в различных культурах.
в массовую долю нитрата в
анализируемой пробе. Данные таблицы составлены с учетом среднего содержания
влаги в различных культурах.
Если при анализе использовался сок, то содержание нитратов в пробе в мг/кг определяют по табл. 24 - 27, которые составлены с учетом эмпирических коэффициентов пересчета.
Если при использовании сока был получен результат по содержанию нитратов превышающий допустимый уровень (см. табл. 21), то для окончательного суждения о качестве продукции следует повторить анализ с использованием навески измельченного материала.
При проведении арбитражных анализов на содержание нитратов в капусте, картофеле, свекле, моркови и зеленых овощах использование сока не допускается.
Оценка результатов испытания. Вычисления проводят до целых чисел, мг/кг. За окончательный результат, выполненный в одной лаборатории, принимают среднее арифметическое (Х) результатов двух параллельных определений. Допустимое расхождение между двумя параллельными определениями, выполняемыми в одной лаборатории, зависит от уровня концентрации и при Р = 0,95 не должно превышать значений сходимости (r), указанных в табл. 28.
Допустимое расхождение между результатами испытаний, выполненных в разных лабораториях, зависит от уровня концентраций и при Р = 0,95 не должно превышать значений воспроизводимости (R), указанных в табл. 28.
Для получения величин сходимости и воспроизводимости для концентраций нитратов, отличных от указанных в табл. 28, можно использовать аппроксимирующие коррелятивные уравнения для зависимости характеристик точности определения нитратов от концентрации (Х, мг/кг)
|
r = 13,8 + 0,08X, R = 0,354X. |
(64) |
При сравнении аналитических результатов с величиной ПДК (табл. 29) в соответствии с рекомендациями Международного стандарта JSC № 5725, 1981, используется величина допустимого критического отклонения СrД, равная:
|
|
(65) |
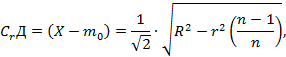
где:
Х - средняя концентрация, полученная в одной лаборатории при
n - параллельных определений;
m0 - величина ПДК.
Если отклонение обнаруженной концентрации от ПДК не превышает значений СrД, представленных в табл. 28 при доверительной вероятности Р = 0,95, можно принять, что определяемая концентрация сопоставима с ПДК; если отклонение превышает СrД, следует считать обнаруженную концентрацию нитратов в продукте не соответствующей уровню ПДК.
Перевод
значений ![]() в массовую
долю нитратов в NO-3
(млн.-1, мг/кг) при анализе вытяжки из картофеля, столовой свеклы,
лука-репки, винограда (H:V = 1:5)
в массовую
долю нитратов в NO-3
(млн.-1, мг/кг) при анализе вытяжки из картофеля, столовой свеклы,
лука-репки, винограда (H:V = 1:5)
|
|
Сотые
доли |
|||||||||
|
.00 |
.01 |
.02 |
.03 |
.04 |
.05 |
.06 |
.07 |
.08 |
.09 |
|
|
1,6 |
9033 |
8827 |
8626 |
8430 |
8238 |
8050 |
7867 |
7688 |
7513 |
7342 |
|
1,7 |
7175 |
7012 |
6852 |
6696 |
6544 |
6375 |
6249 |
6107 |
5968 |
5832 |
|
1,8 |
5699 |
5570 |
5443 |
5319 |
5198 |
5079 |
4964 |
4851 |
4740 |
4633 |
|
1,9 |
4527 |
4424 |
4323 |
4225 |
4129 |
4035 |
3943 |
3853 |
3765 |
3680 |
|
2,0 |
3596 |
3514 |
3434 |
3356 |
3280 |
3205 |
3132 |
3061 |
2991 |
2923 |
|
2,1 |
2856 |
2791 |
2728 |
2666 |
2605 |
2546 |
2488 |
2431 |
2376 |
2322 |
|
2,2 |
2269 |
2217 |
2167 |
2117 |
2069 |
2022 |
1976 |
1931 |
1887 |
1844 |
|
2,3 |
1802 |
1761 |
1721 |
1682 |
1644 |
1606 |
1570 |
1534 |
1499 |
1465 |
|
2,4 |
1432 |
1399 |
1367 |
1336 |
1306 |
1276 |
1247 |
1218 |
1191 |
1164 |
|
2,5 |
1137 |
1111 |
1086 |
1061 |
1037 |
1013 |
990 |
968 |
946 |
924 |
|
2,6 |
903 |
883 |
863 |
843 |
824 |
805 |
787 |
769 |
751 |
734 |
|
2,7 |
717 |
701 |
685 |
670 |
654 |
639 |
625 |
611 |
597 |
583 |
|
2,8 |
570 |
557 |
544 |
532 |
520 |
508 |
496 |
485 |
474 |
463 |
|
2,9 |
453 |
442 |
432 |
422 |
413 |
403 |
394 |
385 |
377 |
368 |
|
3,0 |
366 |
351 |
343 |
336 |
328 |
320 |
313 |
306 |
299 |
292 |
|
3,1 |
286 |
279 |
273 |
267 |
261 |
255 |
249 |
243 |
238 |
232 |
|
3,2 |
227 |
222 |
217 |
212 |
207 |
202 |
198 |
193 |
189 |
184 |
|
3,3 |
180 |
176 |
172 |
168 |
164 |
161 |
157 |
153 |
150 |
146 |
|
3,4 |
143 |
140 |
137 |
134 |
131 |
128 |
125 |
122 |
119 |
116 |
|
3,5 |
114 |
111 |
109 |
106 |
104 |
101 |
99 |
97 |
95 |
92 |
|
3,6 |
90,3 |
88,3 |
86,3 |
84,3 |
82,4 |
80,5 |
78,7 |
76,9 |
75,1 |
73,4 |
|
3,7 |
70,7 |
70,1 |
68,5 |
70,0 |
65,4 |
63,9 |
62,5 |
61,1 |
59,7 |
58,3 |
|
3,8 |
57,0 |
55,7 |
54,4 |
53,2 |
52,0 |
50,8 |
49,6 |
48,5 |
47,4 |
46,3 |
|
3,9 |
45,4 |
44,2 |
43,2 |
42,2 |
41,3 |
40,3 |
39,4 |
38,5 |
37,7 |
36,8 |
|
4,0 |
36,0 |
35,1 |
34,3 |
33,6 |
32,8 |
32,0 |
31,3 |
30,6 |
29,9 |
29,2 |
Перевод
значений ![]() в массовую
долю нитратов в NO-3
(млн.-1, мг/кг) при анализе вытяжки из капусты белокочанной,
моркови, томатов, огурцов, лука-перо, дыни, арбузов, тыквы, перца сладкого, кабачков,
зеленых культур, яблок, груш (H:V = 1:5)
в массовую
долю нитратов в NO-3
(млн.-1, мг/кг) при анализе вытяжки из капусты белокочанной,
моркови, томатов, огурцов, лука-перо, дыни, арбузов, тыквы, перца сладкого, кабачков,
зеленых культур, яблок, груш (H:V = 1:5)
|
|
Сотые
доли |
|||||||||
|
.00 |
.01 |
.02 |
.03 |
.04 |
.05 |
.06 |
.07 |
.08 |
.09 |
|
|
1,6 |
9188 |
8979 |
8775 |
8575 |
8380 |
8189 |
8003 |
7821 |
7643 |
7469 |
|
1,7 |
7299 |
7133 |
6970 |
6812 |
6656 |
6505 |
6357 |
6212 |
6071 |
5933 |
|
1,8 |
5798 |
5666 |
5537 |
5411 |
5287 |
5167 |
5049 |
4935 |
4822 |
4712 |
|
1,9 |
4605 |
4500 |
4398 |
4298 |
4200 |
4104 |
4011 |
3920 |
3830 |
3743 |
|
2,0 |
3658 |
3575 |
3493 |
3414 |
3336 |
3260 |
3186 |
3113 |
3043 |
2973 |
|
2,1 |
2906 |
2840 |
2775 |
2712 |
2650 |
2590 |
2531 |
2473 |
2417 |
2362 |
|
2,2 |
2308 |
2256 |
2204 |
2154 |
2105 |
2057 |
2010 |
1964 |
1920 |
1876 |
|
2,3 |
1833 |
1792 |
1751 |
1711 |
1672 |
1634 |
1597 |
1560 |
1525 |
1490 |
|
2,4 |
1456 |
1423 |
1391 |
1359 |
1328 |
1298 |
1268 |
1239 |
1211 |
1184 |
|
2,5 |
1157 |
1130 |
1105 |
1080 |
1055 |
1031 |
1007 |
985 |
962 |
940 |
|
2,6 |
919 |
898 |
877 |
858 |
838 |
819 |
800 |
782 |
764 |
747 |
|
2,7 |
730 |
713 |
697 |
681 |
666 |
650 |
636 |
621 |
607 |
593 |
|
2,8 |
580 |
567 |
554 |
541 |
529 |
517 |
505 |
493 |
482 |
471 |
|
2,9 |
461 |
450 |
440 |
430 |
420 |
410 |
401 |
392 |
383 |
374 |
|
3,0 |
366 |
357 |
349 |
341 |
334 |
326 |
319 |
311 |
304 |
297 |
|
3,1 |
291 |
284 |
277 |
271 |
265 |
259 |
253 |
247 |
242 |
236 |
|
3,2 |
231 |
226 |
220 |
215 |
210 |
206 |
201 |
196 |
192 |
188 |
|
3,3 |
183 |
179 |
175 |
171 |
167 |
163 |
166 |
156 |
152 |
149 |
|
3,4 |
146 |
142 |
139 |
136 |
130 |
130 |
127 |
124 |
121 |
118 |
|
3,5 |
116 |
113 |
110 |
108 |
105 |
103 |
101 |
98 |
96 |
94 |
|
3,6 |
91,9 |
89,8 |
87,7 |
85,8 |
83,8 |
81,9 |
80,0 |
78,2 |
76,4 |
74,7 |
|
3,7 |
73,0 |
71,3 |
69,7 |
68,1 |
66,6 |
65,0 |
63,6 |
62,1 |
60,7 |
59,3 |
|
3,8 |
58,0 |
56,7 |
55,4 |
54,1 |
52,9 |
51,7 |
50,5 |
49,3 |
48,2 |
47,1 |
|
3,9 |
46,1 |
45,0 |
44,0 |
43,0 |
42,0 |
41,0 |
40,1 |
39,2 |
38,3 |
37,4 |
|
4,0 |
36,6 |
35,7 |
34,9 |
34,1 |
33,4 |
32,6 |
31,9 |
31,1 |
30,4 |
29,7 |
Перевод
значений ![]() в массовую
долю нитратов в NO-3
(млн.-1, мг/кг) при анализе сока картофеля и лука-репки (V
сока:V экстрагирующего
раствора = 1:5)
в массовую
долю нитратов в NO-3
(млн.-1, мг/кг) при анализе сока картофеля и лука-репки (V
сока:V экстрагирующего
раствора = 1:5)
|
|
Сотые
доли |
|||||||||
|
.00 |
.01 |
.02 |
.03 |
.04 |
.05 |
.06 |
.07 |
.08 |
.09 |
|
|
1,6 |
7475 |
7305 |
7139 |
6976 |
6818 |
6662 |
6511 |
6363 |
6218 |
6076 |
|
1,7 |
5938 |
5803 |
5671 |
5542 |
5415 |
5292 |
5172 |
5054 |
4939 |
4827 |
|
1,8 |
4717 |
4609 |
4504 |
4402 |
4302 |
4204 |
4108 |
4015 |
3923 |
3834 |
|
1,9 |
3747 |
3661 |
3578 |
3496 |
3417 |
3339 |
3263 |
3189 |
3116 |
3045 |
|
2,0 |
2976 |
2908 |
2842 |
2777 |
2714 |
2652 |
2592 |
2533 |
2475 |
2419 |
|
2,1 |
2364 |
2310 |
2258 |
2206 |
2156 |
2107 |
2059 |
2012 |
1966 |
1921 |
|
2,2 |
1878 |
1835 |
1793 |
1752 |
1713 |
1674 |
1635 |
1598 |
1562 |
1526 |
|
2,3 |
1492 |
1458 |
1424 |
1392 |
1360 |
1329 |
1299 |
1270 |
1241 |
1212 |
|
2,4 |
1185 |
1158 |
1131 |
1106 |
1081 |
1056 |
1032 |
1008 |
985 |
963 |
|
2,5 |
941 |
920 |
899 |
878 |
858 |
839 |
820 |
801 |
783 |
765 |
|
2,6 |
748 |
731 |
714 |
698 |
682 |
666 |
651 |
636 |
622 |
608 |
|
2,7 |
594 |
580 |
567 |
554 |
542 |
529 |
517 |
505 |
494 |
483 |
|
2,8 |
472 |
461 |
450 |
440 |
430 |
420 |
411 |
401 |
392 |
383 |
|
2,9 |
375 |
366 |
358 |
350 |
342 |
334 |
326 |
319 |
312 |
305 |
|
3,0 |
298 |
291 |
284 |
278 |
271 |
265 |
259 |
253 |
248 |
242 |
|
3,1 |
236 |
231 |
226 |
221 |
216 |
211 |
206 |
201 |
197 |
192 |
|
3,2 |
188 |
183 |
179 |
175 |
171 |
167 |
164 |
160 |
156 |
153 |
|
3,3 |
149 |
146 |
142 |
139 |
136 |
133 |
130 |
127 |
124 |
121 |
|
3,4 |
118 |
116 |
113 |
111 |
108 |
106 |
103 |
101 |
99 |
96 |
|
3,5 |
94 |
92 |
90 |
88 |
86 |
84 |
82 |
80 |
78 |
76 |
|
3,6 |
75 |
73 |
71 |
70 |
68 |
67 |
65 |
64 |
62 |
61 |
|
3,7 |
59 |
58 |
57 |
55 |
54 |
53 |
52 |
51 |
49 |
48 |
|
3,8 |
47 |
46 |
45 |
44 |
43 |
42 |
41 |
40 |
39 |
38 |
|
3,9 |
37 |
37 |
36 |
35 |
34 |
33 |
33 |
32 |
31 |
30 |
|
4,0 |
30 |
29 |
28 |
28 |
27 |
27 |
26 |
25 |
25 |
24 |
Перевод
значений ![]() в массовую долю нитратов в NO-3 (млн.-1, мг/кг) при
анализе сока свеклы и моркови (V сока:V экстрагирующего раствора = 1:5)
в массовую долю нитратов в NO-3 (млн.-1, мг/кг) при
анализе сока свеклы и моркови (V сока:V экстрагирующего раствора = 1:5)
|
|
Сотые
доли |
|||||||||
|
.00 |
.01 |
.02 |
.03 |
.04 |
.05 |
.06 |
.07 |
.08 |
.09 |
|
|
1,6 |
7943 |
7762 |
7585 |
7412 |
7244 |
7079 |
6918 |
6760 |
6606 |
6456 |
|
1,7 |
6309 |
6165 |
6025 |
5888 |
5754 |
5623 |
5495 |
5370 |
5248 |
5128 |
|
1,8 |
5011 |
4897 |
4786 |
4677 |
4570 |
4466 |
4363 |
4265 |
4168 |
4073 |
|
1,9 |
3981 |
3890 |
3802 |
3715 |
3630 |
3548 |
3467 |
3388 |
3311 |
3236 |
|
2,0 |
3162 |
3090 |
3020 |
2951 |
2884 |
2818 |
2754 |
2691 |
2630 |
2570 |
|
2,1 |
2512 |
2454 |
2399 |
2344 |
2291 |
2239 |
2188 |
2138 |
2089 |
2042 |
|
2,2 |
1995 |
1950 |
1905 |
1862 |
1820 |
1778 |
1738 |
1698 |
1659 |
1622 |
|
2,3 |
1585 |
1549 |
1513 |
1479 |
1445 |
1412 |
1380 |
1349 |
1318 |
1288 |
|
2,4 |
1259 |
1230 |
1202 |
1175 |
1148 |
1122 |
1096 |
1071 |
1047 |
1023 |
|
2,5 |
1000 |
977 |
955 |
933 |
912 |
891 |
871 |
851 |
832 |
813 |
|
2,6 |
794 |
776 |
759 |
741 |
724 |
708 |
692 |
676 |
661 |
646 |
|
2,7 |
631 |
617 |
603 |
589 |
575 |
562 |
549 |
537 |
525 |
513 |
|
2,8 |
501 |
490 |
479 |
468 |
457 |
447 |
436 |
427 |
417 |
407 |
|
2,9 |
398 |
389 |
380 |
372 |
363 |
355 |
347 |
339 |
331 |
324 |
|
3,0 |
316 |
309 |
302 |
295 |
288 |
282 |
275 |
269 |
263 |
257 |
|
3,1 |
251 |
245 |
240 |
234 |
229 |
224 |
219 |
214 |
209 |
204 |
|
3,2 |
200 |
195 |
191 |
186 |
182 |
178 |
174 |
170 |
166 |
162 |
|
3,3 |
158 |
155 |
151 |
148 |
145 |
141 |
138 |
135 |
132 |
129 |
|
3,4 |
126 |
123 |
120 |
117 |
115 |
112 |
110 |
107 |
105 |
102 |
|
3,5 |
100 |
98 |
95 |
93 |
91 |
89 |
87 |
85 |
83 |
81 |
|
3,6 |
79 |
78 |
76 |
74 |
72 |
71 |
69 |
68 |
66 |
65 |
|
3,7 |
63 |
62 |
60 |
59 |
58 |
56 |
55 |
54 |
52 |
51 |
|
3,8 |
50 |
49 |
48 |
47 |
46 |
45 |
44 |
43 |
42 |
41 |
|
3,9 |
40 |
39 |
38 |
37 |
36 |
35 |
35 |
34 |
33 |
32 |
|
4,0 |
32 |
31 |
30 |
30 |
29 |
28 |
28 |
27 |
26 |
26 |
Перевод
значений ![]() в массовую
долю нитратов в NO-3
(млн.-1, мг/кг) при анализе сока капусты белокочанной, перца сладкого,
винограда (V сока:V экстрагирующего раствора = 1:5)
в массовую
долю нитратов в NO-3
(млн.-1, мг/кг) при анализе сока капусты белокочанной, перца сладкого,
винограда (V сока:V экстрагирующего раствора = 1:5)
|
|
Сотые
доли |
|||||||||
|
.00 |
.01 |
.02 |
.03 |
.04 |
.05 |
.06 |
.07 |
.08 |
.09 |
|
|
1,6 |
8410 |
8218 |
8031 |
7848 |
7670 |
7495 |
7325 |
7158 |
6995 |
6836 |
|
1,7 |
6680 |
6528 |
6379 |
6234 |
6092 |
5954 |
5818 |
5686 |
5556 |
5430 |
|
1,8 |
5306 |
5185 |
5667 |
4952 |
4839 |
4729 |
4622 |
4516 |
4416 |
4313 |
|
1,9 |
4215 |
4119 |
4025 |
3934 |
3844 |
3757 |
3671 |
3587 |
3506 |
3426 |
|
2,0 |
3348 |
3272 |
3197 |
3125 |
3053 |
2984 |
2916 |
2850 |
2785 |
2721 |
|
2,1 |
2659 |
2599 |
2540 |
2482 |
2425 |
2370 |
2316 |
2264 |
2212 |
2162 |
|
2,2 |
2112 |
2064 |
2017 |
1971 |
1927 |
1883 |
1840 |
1798 |
1757 |
1717 |
|
2,3 |
1678 |
1640 |
1602 |
1566 |
1530 |
1495 |
1461 |
1428 |
1396 |
1364 |
|
2,4 |
1333 |
1303 |
1273 |
1244 |
1216 |
1188 |
1161 |
1134 |
1109 |
1083 |
|
2,5 |
1059 |
1035 |
1010 |
988 |
966 |
944 |
922 |
901 |
881 |
861 |
|
2,6 |
841 |
822 |
803 |
785 |
767 |
750 |
732 |
716 |
699 |
684 |
|
2,7 |
668 |
653 |
638 |
623 |
609 |
595 |
582 |
569 |
556 |
543 |
|
2,8 |
531 |
519 |
507 |
495 |
484 |
473 |
462 |
452 |
441 |
431 |
|
2,9 |
421 |
412 |
403 |
393 |
384 |
376 |
367 |
359 |
351 |
343 |
|
3,0 |
335 |
327 |
320 |
312 |
305 |
298 |
292 |
285 |
278 |
272 |
|
3,1 |
266 |
260 |
254 |
248 |
243 |
237 |
232 |
226 |
221 |
216 |
|
3,2 |
211 |
206 |
202 |
197 |
193 |
188 |
184 |
180 |
176 |
172 |
|
3,3 |
168 |
164 |
160 |
157 |
153 |
150 |
146 |
143 |
140 |
136 |
|
3,4 |
133 |
130 |
127 |
124 |
122 |
119 |
116 |
113 |
111 |
108 |
|
3,5 |
106 |
103 |
101 |
99 |
97 |
94 |
92 |
90 |
88 |
86 |
|
3,6 |
84 |
82 |
80 |
78 |
77 |
75 |
73 |
72 |
70 |
68 |
|
3,7 |
67 |
65 |
64 |
62 |
61 |
60 |
58 |
57 |
56 |
54 |
|
3,8 |
53 |
52 |
51 |
50 |
48 |
47 |
46 |
45 |
44 |
43 |
|
3,9 |
42 |
41 |
40 |
39 |
38 |
38 |
37 |
36 |
35 |
34 |
|
4,0 |
33 |
33 |
32 |
31 |
31 |
30 |
29 |
28 |
28 |
27 |
Перевод
значений ![]() в массовую долю нитратов NO-3 (млн.-1, мг/кг) при
анализе сока огурцов, томатов, кабачков, дынь, арбузов, тыквы, яблок, груш (V
сока:V экстрагирующего раствора = 1:5)
в массовую долю нитратов NO-3 (млн.-1, мг/кг) при
анализе сока огурцов, томатов, кабачков, дынь, арбузов, тыквы, яблок, груш (V
сока:V экстрагирующего раствора = 1:5)
|
|
Сотые
доли |
|||||||||
|
.00 |
.01 |
.02 |
.03 |
.04 |
.05 |
.06 |
.07 |
.08 |
.09 |
|
|
1,6 |
8877 |
8675 |
8477 |
8285 |
8096 |
7912 |
7732 |
7556 |
7384 |
7216 |
|
1,7 |
7051 |
6891 |
6734 |
6581 |
6431 |
6284 |
6141 |
6002 |
5865 |
5731 |
|
1,8 |
5601 |
5474 |
5349 |
5227 |
5108 |
4992 |
4878 |
4767 |
4659 |
4553 |
|
1,9 |
4449 |
4348 |
4249 |
4152 |
4058 |
3965 |
3875 |
3787 |
3701 |
3616 |
|
2,0 |
3534 |
3454 |
3375 |
3298 |
3223 |
3150 |
3078 |
3008 |
2939 |
2873 |
|
2,1 |
2807 |
2743 |
2681 |
2620 |
2560 |
2502 |
2445 |
2389 |
2335 |
2282 |
|
2,2 |
2230 |
2179 |
2129 |
2081 |
2034 |
1987 |
1942 |
1898 |
1855 |
1812 |
|
2,3 |
1771 |
1731 |
1691 |
1653 |
1615 |
1579 |
1543 |
1508 |
1473 |
1440 |
|
2,4 |
1407 |
1375 |
1344 |
1313 |
1283 |
1254 |
1225 |
1197 |
1170 |
1144 |
|
2,5 |
1118 |
1092 |
1067 |
1043 |
1019 |
996 |
973 |
951 |
930 |
908 |
|
2,6 |
888 |
867 |
848 |
828 |
810 |
791 |
773 |
756 |
738 |
722 |
|
2,7 |
705 |
689 |
673 |
658 |
643 |
628 |
614 |
600 |
586 |
573 |
|
2,8 |
560 |
547 |
535 |
523 |
511 |
499 |
488 |
477 |
466 |
455 |
|
2,9 |
445 |
435 |
425 |
415 |
406 |
397 |
387 |
379 |
370 |
362 |
|
3,0 |
353 |
345 |
337 |
330 |
322 |
315 |
308 |
301 |
294 |
287 |
|
3,1 |
281 |
274 |
268 |
262 |
256 |
250 |
244 |
239 |
233 |
228 |
|
3,2 |
223 |
218 |
213 |
208 |
203 |
199 |
194 |
190 |
185 |
181 |
|
3,3 |
177 |
173 |
169 |
165 |
162 |
158 |
154 |
151 |
147 |
144 |
|
3,4 |
141 |
137 |
134 |
131 |
128 |
125 |
123 |
120 |
117 |
114 |
|
3,5 |
112 |
109 |
107 |
104 |
102 |
100 |
97 |
95 |
93 |
91 |
|
3,6 |
89 |
87 |
85 |
83 |
81 |
79 |
77 |
76 |
74 |
72 |
|
3,7 |
71 |
69 |
67 |
66 |
64 |
63 |
61 |
60 |
59 |
57 |
|
3,8 |
56 |
55 |
53 |
52 |
51 |
50 |
49 |
48 |
47 |
46 |
|
3,9 |
44 |
43 |
42 |
42 |
41 |
40 |
39 |
38 |
37 |
36 |
|
4,0 |
35 |
35 |
34 |
33 |
32 |
31 |
31 |
30 |
29 |
29 |
Внутрилабораторная сходимость (r), межлабораторная воспроизводимость (R) и допустимое критическое отклонение от ПДК (СrД) для ионометрического метода определения нитратов при различных уровнях концентраций (Х) в растениеводческой продукции (мг/кг)
|
X, мг/кг |
r, мг/кг |
R, мг/кг |
СrД при n = 2 |
|
50 |
18 |
18 |
9 |
|
60 |
19 |
21 |
11 |
|
80 |
20 |
28 |
17 |
|
90 |
21 |
32 |
20 |
|
100 |
22 |
35 |
22 |
|
150 |
26 |
53 |
40 |
|
200 |
30 |
71 |
48 |
|
250 |
34 |
88 |
60 |
|
300 |
38 |
106 |
72 |
|
400 |
46 |
142 |
98 |
|
500 |
55 |
177 |
123 |
|
600 |
62 |
212 |
147 |
|
750 |
75 |
265 |
184 |
|
800 |
78 |
283 |
196 |
|
900 |
86 |
319 |
221 |
|
1000 |
95 |
354 |
251 |
|
1400 |
136 |
496 |
371 |
|
2000 |
177 |
708 |
487 |
|
2500 |
218 |
885 |
618 |
|
3000 |
258 |
1062 |
742 |
2.10.2. Фотометрический метод определения нитритов и нитратов
Метод применяют при анализе всех видов продукции. Метод определения нитритов основан на экстрагировании нитритов водой, очистке экстракта и фотометрическом измерении интенсивности окраски, образующейся при взаимодействии нитрит-иона (NO-2) с ароматическими аминами. Нижний предел обнаружения нитрит-иона в колориметрируемом растворе - 0,02 мкг/см3, нижний предел надежного определения в анализируемой пробе - 0,5 мг/кг.
Метод определения нитратов основан на экстрагировании их водой, очистке экстракта, количественном восстановлении нитратов в нитриты на кадмиевой колонке с последующим фотометрическим измерением интенсивности окраски азосоединения, образующегося при взаимодействии нитритов с ароматическими аминами. Нижний предел обнаружения нитрат-иона в колориметрируемом растворе - 0,03 мг/см3, нижний предел надежного определения в анализируемой пробе - 1,5 мг/кг.
На выбор исследователя предлагаются два равноценных варианта определения нитритов и нитратов.
Вариант I содержит следующие этапы работы: отбор и подготовка проб, приготовление растворов для испытаний, подготовка кадмиевой колонки к работе, построение градуировочного графика для определения нитратов, проведение испытаний проб на содержание нитритов и нитратов, обработка результатов анализа.
Вариант II в основном аналогичен первому и отличается тем, что стадия "построения градуировочного графика для определения нитратов" заменяется стадией "проверка восстановительной способности колонки", при этом расчет содержания нитратов проводится на основе градуировочного графика по нитритам.
Вариант I
Определение нитратов
Аппаратура, материалы, реактивы. Весы лабораторные; ареометры; баня водяная с терморегулятором; спектрофотометр или фотоэлектроколориметр различных марок; колбы конические вместимостью 50, 100, 200, 250, 300 см3; колбы мерные вместимостью 50, 100, 200, 500, 1000 см3; цилиндры мерные вместимостью 25, 50, 100, 250 см3; стаканы химические вместимостью 50, 500, 1000 см3; пипетки (бюретки, дозаторы) вместимостью 1, 5, 10, 20 см3; воронки стеклянные; палочки стеклянные; фильтры обеззоленные (перед употреблением фильтры необходимо промыть не менее 5 раз дистиллированной водой и высушить в сушильном шкафу в течение 0,5 часа при 70 - 80 °C); бумага универсальная индикаторная; цинк металлический гранулированный (или в палочках 150×8 мм); сульфат кадмия; сульфат цинка 7-водный; гексацианоферрат (II) калия 3-водный; нитрат калия; кислота соляная плотностью 1,19 г/см3; аммиак водный плотностью 0,9 г/см3; сульфаниламид; сульфаниловая кислота; N-(1-нафтил)этилендиамин дигидрохлорид; 1-нафтиламин; кислота уксусная; вода дистиллированная; нитрит натрия; тетраборат натрия; гидроксид натрия.
Подготовка к испытанию. Приготовление растворов для осаждения белков. Раствор сульфата цинка: 535 г сульфата цинка, взвешенного с точностью до 0,1 г, растворяют дистиллированной водой в мерной колбе вместимостью 1000 см3 и доводят объем раствора до метки. Раствор гексацианоферрата (II) калия: 172 г гексацианоферрата (II) калия, взвешенного с точностью до 0,1 г, растворяют дистиллированной водой в мерной колбе вместимостью 1000 см3 и доводят объем раствора до метки. Насыщенный раствор тетрабората натрия: 50 г тетрабората натрия, взвешенного с точностью до 0,1 г, растворяют дистиллированной водой с температурой 50 ± 2 °C в мерной колбе вместимостью 1000 см3, затем охлаждают до температуры 20 ± 2 °C и доводят объем до метки. Раствор гидроксида натрия концентрацией 1 моль/дм3. Аммиачный буфер: 50 см3 соляной кислоты плотностью 1,19 г/см3 растворяют в 600 см3 дистиллированной воды, добавляют 135 см3 концентрированного водного аммиака плотностью 0,9 г/см3, перемешивают. Проверяют pH раствора и при необходимости доводят pH до 9,6 - 9,7, доводят дистиллированной водой до метки. Раствор соляной кислоты концентрацией 0,1 моль/дм3 (0,1 н): 7,8 см3 соляной кислоты плотностью 1,19 г/см3 растворяют дистиллированной водой в мерной колбе вместимостью 1000 см3 и доводят объем раствора до метки.
Приготовление растворов для проведения цветной реакции:
1) раствор сульфаниламида: 1,0 г сульфаниламида, взвешенного с точностью до 0,1 г, помещают в мерную колбу вместимостью 250 см3, добавляют 200 см3 дистиллированной воды и 10 см3 концентрированной соляной кислоты. Смесь выдерживают на кипящей водяной бане до полного растворения сульфаниламида, затем раствор охлаждают и доводят до метки дистиллированной водой. Если необходимо, фильтруют;
2) раствор N-(1-нафтил)этилендиамин дигидрохлорида (НЭДА): 0,1 г НЭДА, взвешенного с точностью до 0,1 г, растворяют дистиллированной водой в мерной колбе вместимостью 50 см3 и доводят объем раствора до метки. Растворы сульфаниламида и НЭДА хранят в холодильнике в склянке из темного стекла с притертой пробкой не более 2 недель;
3) раствор сульфаниловой кислоты: 1,5 г сульфаниловой кислоты, взвешенной с точностью до 0,1 г, растворяют в 200 см3 горячей дистиллированной воды и после охлаждения добавляют 50 см3 ледяной уксусной кислоты;
4) раствор 1-нафтиламина: 1,2 г 1-нафтиламина, взвешенного с точностью до 0,1 г, растворяют в 50 см3 ледяной уксусной кислоты и доводят объем раствора дистиллированной водой до 200 см3. Растворы сульфаниловой кислоты и 1-нафтиламина хранят в холодильнике в склянке из темного стекла с притертой пробкой не более 2 недель;
5) раствор соляной кислоты: 445 см3 соляной кислоты (плотность 1,19 г/см3) растворяют дистиллированной водой в мерной колбе вместимостью 1 дм3 и доводят объем раствора до метки.
Приготовление основного стандартного раствора нитратов: 1,630 г нитрата калия, перекристаллизованного из воды и высушенного до постоянной массы при температуре 110 - 120 °C, взвешивают с точностью до 0,001 г, растворяют дистиллированной водой в мерной колбе вместимостью 1000 см3 и доводят объем раствора до метки. Хранят в склянке с притертой пробкой в холодильнике до 6 месяцев.
Рабочий раствор нитратов: 10 см3 с точностью до 1 см 3основного стандартного раствора нитратов помещают в мерную колбу вместимостью 1 дм3 и доводят дистиллированной водой до метки. 1 см3 раствора содержит 10 мкг NO-3. Хранят в склянке с притертой пробкой в холодильнике не более 1 месяца.
Подготовка кадмиевой колонки. Приготовление пористого кадмия. В химическом стакане вместимостью 1 дм3 растворяют 20 г сернокислого кадмия в 500 см3 дистиллированной воды и помещают 1 - 2 палочки металлического цинка (возможно использование гранулированного цинка). По мере образования кадмия его удаляют при помощи шпателя в другой стакан с дистиллированной водой. Собранный кадмий декантацией промывают несколько раз дистиллированной водой, используя не менее 2 дм3, суспензию кадмия переносят в гомогенизатор и измельчают до получения частиц диаметром 0,3 - 0,8 мм. Содержимое гомогенизатора переносят в химический стакан, жидкость декантируют и кадмий многократно промывают (5 - 6 раз), используя по 100 - 200 см3 0,1 моль/дм3 (0,1 н) раствора соляной кислоты. При этом декантацией отделяют мелкую фракцию кадмия. Хранят кадмий под слоем 0,1 моль/дм3 (0,1 н) раствора соляной кислоты. В день анализа кадмий с целью удаления пузырьков газа перемешивают. Жидкость декантируют и кадмий промывают несколько раз дистиллированной водой до нейтральной реакции по универсальной индикаторной бумаге.
При подготовке кадмиевой колонки к работе собирают систему согласно рис. 2. Сначала ее полностью заполняют водой и при открытом кране вносят суспензию пористого кадмия до тех пор, пока высота слоя кадмия не достигнет 130 - 150 мм. Поверхность кадмия в колонке должна всегда быть покрыта жидкостью.
Перед началом работы и после каждого анализа кадмиевую колонку промывают последовательно 25 см3 0,1 моль/дм3 (0,1 н) раствора соляной кислоты, 50 см3 дистиллированной воды и 25 см3 разбавленного (1:9) аммиачного буфера.
Рис. 2. Принципиальная схема кадмиевои колонки
Построение градуировочного графика для определения нитратов: в стаканы или колбы на 50 см3 вносят пипеткой 1,0; 2,0; 4,0; 8,0; 10,0 см3 рабочего раствора с. 124), добавляют дистиллированную воду до объема 20 см3, 5 см3 аммиачного буфера и перемешивают. Для приготовления контрольного раствора в стакан или колбу на 50 см3 вносят пипеткой 20 см3 дистиллированной воды и 5 см3 аммиачного буфера.
Через предварительно промытую кадмиевую колонку пропускают со скоростью 5 см/мин3 сначала один из полученных растворов нитратов, затем 25 см3 дистиллированной воды, собирая элюат в мерную колбу вместимостью 50 см3, и доводят объем до метки.
25 см3 полученного элюата помещают в мерную колбу вместимостью 50 см3 и проводят цветную реакцию, для чего в каждую колбу добавляют по 5 см3 раствора сульфаниламида (п. 1) и по 1 см3 раствора соляной кислоты (п. 5), перемешивают и оставляют на 5 мин. Затем добавляют по 1 см3 раствора НЭДА (п. 2), доводят объем до метки дистиллированной водой, перемешивают и через 10 мин колориметрируют в кюветах толщиной 10 мм при длине волны 538 нм по отношению к контрольному раствору.
В случае использования сульфаниловой кислоты и 1-нафтиламина в колбы добавляют по 1 см3 раствора сульфаниловой кислоты (п. 3), 3 см3 раствора соляной кислоты (п. 5), перемешивают и оставляют на 5 мин при комнатной температуре. Затем добавляют по 1 см3 раствора 1-нафтиламина (п. 4), перемешивают, доводят дистиллированной водой до метки и колориметрируют через 2 часа.
По полученным данным строят градуировочный график, откладывая по оси ординат значение оптической плотности, а по оси абсцисс - значения концентрации нитрат-иона: 0,1; 0,2; 0,4; 0,8 и 1,0 мкг/см3 колориметрируемого раствора.
Проведение испытания. Пробы продукции тщательно измельчают с помощью терки, мясорубки или гомогенизатора. Из подготовленной пробы отбирают навеску в количестве 20 г (при низких до 50 мг/кг содержаниях нитратов) или 10 г (в остальных случаях) с точностью до 0,1 г. Навеску продукта переносят в 200 - 300 см3 коническую колбу, добавляют 100 см3 теплой дистиллированной воды (60 - 65 °C) и тщательно перемешивают. Колбу помещают на водяную баню (60 - 65 °C), для осаждения белков вносят 5 см3 насыщенного раствора натрия тетрабората, 5 см3 раствора сульфата цинка и 5 см3 раствора гексацианоферрата (II) калия, перемешивая содержимое после добавления каждого реактива. Выдерживают при этой температуре 15 мин, затем охлаждают до комнатной температуры. Содержимое колбы количественно переносят в мерную колбу на 200 см3, ополаскивают два раза по 10 - 15 см3 теплой дистиллированной водой и объединяют с основной смесью; объем доводят водой до метки и фильтруют через бумажный фильтр (фильтрат I). Фильтрат I служит для определения нитратов и нитритов.
При получении мутного фильтрата в качестве осадителей можно использовать 5 см3 раствора сульфата цинка и 10 см3 1 моль/дм3 (1 н) раствора гидроксида натрия.
Перед началом анализа необходимо оценить наличие нитратов и нитритов во всех используемых реактивах (холостая проба), для чего проводят все операции без внесения продукта. Для этого в мерную колбу на 200 см3 вносят 100 см3 дистиллированной воды, по 5 см3 насыщенного раствора натрия тетрабората, раствора сульфата цинка и раствора гексацианоферрата (II) калия или 5 см3 раствора сульфата цинка и 10 см3 раствора гидроксида натрия концентрацией 1 моль/дм3 (1 н), перемешивают содержимое колбы после добавления каждого реактива, затем объем доводят водой до метки и фильтруют через бумажный фильтр. В фильтрате (холостая проба) определяют содержание нитратов и нитритов.
Если содержание нитратов и нитритов в "холостой пробе" превышает 0,03 мкг/дм3, то используемые в опыте реактивы следует подвергнуть дополнительной очистке (перегонке, перекристаллизации) или заменить.
Определение содержания нитратов в образце. В стакан или колбу вместимостью 50 см3 вносят от 1 до 40 см3 фильтрата I в зависимости от предполагаемого содержания нитратов в анализируемой продукции. При ожидаемой концентрации от 0 до 100 мг/кг NO-3 объем фильтрата, используемого для восстановления на колонке, составляет 40 см3, 100 - 300 мг/кг NO-3 - 20 см3, 300 - 600 мг/кг NO-3 - 10 см3, 600 - 1000 мг/кг NO-3 - 5 см3, свыше 1000 мг/кг NO-3 - 2 см3. К указанному раствору добавляют 5 см3 аммиачного буфера, перемешивают, пропускают через кадмиевую колонку со скоростью 5 см3/мин сначала полученный раствор образца, затем колонку промывают дистиллированной водой, собирая элюат в мерную колбу вместимостью 100 см3 и доводят объем до метки.
25 см3 полученного элюата помещают в мерную колбу вместимостью 100 см3 и проводят цветную реакцию (с. 126), увеличив объем реактивов для цветной реакции в 2 раза. Величину оптической плотности раствора определяют по отношению к контрольной пробе.
Для получения контрольной пробы в мерную колбу вместимостью 100 см3 вносят объем фильтрата (0,5 - 10 см3), равный тому объему, который используется для проведения цветной реакции при определении нитратов в образце. К этому фильтрату добавляют 1,0 см3 аммиачного буфера, 50 см3 дистиллированной воды и проводят цветную реакцию, как указано выше.
Пример. Если для восстановления на колонке используется 10 см3 фильтрата I (V2 = 10 см3), объем элюата, собираемого с колонки (V3 = 100 см3), объем элюата, взятый на цветную реакцию (V4 = 25 см3), а объем колориметрируемого раствора (V5 = 100 см3), то объем фильтрата I, который следует использовать для получения контрольной пробы, составляет 2,5 см3.
|
Объем фильтрата I, используемый для восстановления на колонке |
Объем фильтрата I, который следует брать для получения контрольной пробы |
|
40 см3 |
10 см3 |
|
20 см3 |
5 см3 |
|
10 см3 |
2,5 см3 |
|
5 см3 |
1,25 см3 |
|
2 см3 |
0,5 см3 |
Обработка результатов анализа. Содержание нитратов (Х), выраженное в мг/кг продукта (в перерасчете на нитрат-ион), вычисляют по формуле
|
|
(66) |
где:
С - концентрация нитрат-иона в колориметрируемом растворе, найденная по градуировочному графику, мкг/см3;
V1 - общий объем экстракта, см3;
V2 - объем фильтрата I, взятый на колонку, см3;
V3 - общий объем элюата, см3;
m - масса навески продукта, г;
V4 - объем элюата, взятый на цветную реакцию, см3;
V5 - объем колориметрируемого раствора, см3.
Определение нитритов
Подготовка к испытанию. Приготовление основного стандартного раствора нитритов: 150 мг перекристаллизованного из воды и высушенного при температуре 110 - 120 °C до постоянной массы нитрата натрия взвешивают с точностью до 0,001 г, помещают в мерную колбу вместимостью 1 дм3, растворяют в дистиллированной воде и доводят объем раствора до метки. Раствор хранят в склянке с притертой пробкой в холодильнике не более одной недели.
Приготовление рабочего раствора нитритов: 10 см3 с точностью до 1 см3 основного стандартного раствора нитритов помещают в мерную колбу вместимостью 500 см3 и доводят дистиллированной водой объем до метки. 1 см3 этого раствора содержит 2,0 мкг NO-2. Раствор готовят в день проведения анализа.
Построение градуировочного графика для определения нитритов: в мерные колбы вместимостью 50 см3 вносят 2,5; 5,0; 10,0; 20,0; 30,0 см3 рабочего раствора нитритов, доводят до 40 см3 дистиллированной водой, в одну мерную колбу вносят 40 см3 дистиллированной воды (контрольный раствор), затем проводят цветную реакцию, для чего в каждую колбу добавляют по 5 см3 раствора сульфаниламида (п. 1) и по 1 см3 раствора соляной кислоты (п. 5), перемешивают и оставляют на 5 минут. Затем добавляют по 1 см3 раствора НЭДА (п. 2), доводят объем до метки дистиллированной водой, перемешивают и через 10 минут колориметрируют в кюветах толщиной 10 мм при длине волны 538 нм по отношению к контрольному раствору.
В случае использования сульфаниловой кислоты и 1-нафтиламина в колбы добавляют по 1 см3 раствора сульфаниловой кислоты (п. 3), 3 см3 раствора соляной кислоты (п. 5), перемешивают и оставляют на 5 минут при комнатной температуре. Затем добавляют по 1 см3 раствора 1-нафтиламина (п. 4), перемешивают, доводят дистиллированной водой до метки и колориметрируют через 2 часа.
По полученным данным строят градуировочный график, откладывая по оси ординат значения оптической плотности; по оси абсцисс - значения концентрации нитрит-иона: 0,1; 0,2; 0,4; 0,8; 1,2 мкг/см3 колориметрируемого раствора.
Определение содержания нитритов в образце. В мерную колбу вместимостью 50 см3 вносят 20 см3 фильтрата I и проводят цветную реакцию, как указано выше, измеряя оптическую плотность раствора относительно смеси, состоящей из 2 объемов фильтрата I и 3 объемов дистиллированной воды.
Обработка результатов испытания. Содержание нитритов (Х), выраженное в мг/кг продукта (в пересчете на нитрит-ион), вычисляют по формуле
|
|
(67) |
где:
С - концентрация нитрит-иона в колориметрируемом растворе, найденная по градуировочному графику, мкг/см3;
V1 - общий объем экстракта, см3;
V2 - объем фильтрата I, взятый на цветную реакцию, см3;
m - масса навески образца, взятая на анализ, г;
V3 - объем колориметрируемого раствора, см3.
Вычисления проводят до целых чисел, мг/кг. За окончательный результат, выполненный в одной лаборатории, принимают среднее арифметическое (Х) результатов двух параллельных определений.
Допустимое расхождение между двумя параллельными определениями, выполненными в одной лаборатории, зависит от уровня концентраций и при Р = 0,95 не должно превышать значений сходимости (r), указанных в табл. 29.
Допустимое расхождение между результатами испытаний, выполненных в разных лабораториях, зависит от уровня концентраций и при Р = 0,95 не должно превышать значений воспроизводимости (R), указанных в табл. 29.
Внутрилабораторная сходимость (r), межлабораторная воспроизводимость (R) и допустимое критическое отклонение от ПДК для фотометрического метода определения нитратов при различных уровнях концентраций (Х) в растениеводческой продукции*
|
X |
r |
R |
СrД при n = 2 |
|
50 |
17 |
36 |
24 |
|
60 |
18 |
41 |
28 |
|
80 |
21 |
52 |
35 |
|
90 |
22 |
57 |
39 |
|
100 |
23 |
62 |
43 |
|
150 |
29 |
86 |
59 |
|
200 |
35 |
109 |
75 |
|
250 |
39 |
134 |
91 |
|
300 |
46 |
153 |
106 |
|
400 |
58 |
193 |
133 |
|
500 |
69 |
238 |
165 |
|
600 |
81 |
269 |
230 |
|
750 |
97 |
332 |
231 |
|
800 |
104 |
341 |
235 |
|
900 |
116 |
376 |
259 |
|
1000 |
126 |
421 |
292 |
|
1400 |
183 |
588 |
407 |
|
2000 |
241 |
745 |
515 |
|
2500 |
298 |
894 |
617 |
|
3000 |
355 |
1041 |
717 |
________________
* Для получения величин r, R, СrД для концентраций, отличных от указанных в табл. 29, можно использовать уравнения:
![]()
При сравнении аналитических результатов с величиной ПДК (табл. 21) используется значение СrД. Если разница обнаруженной концентрации и величина ПДК (или сертификата) не превышает значения СrД, представленной в табл. 29, можно принять, что при доверительной вероятности Р = 0,95 определяемая концентрация сопоставима с ПДК; если полученная разница превышает СrД, обнаруженную концентрацию нитратов в продукте следует считать не соответствующей уровню ПДК.
Вариант II
Все разделы выполняются аналогично варианту I с некоторыми изменениями в части формулы расчета содержания нитратов. Вместо "построения градуировочного графика" для нитратов проводят "проверку восстановительной способности колонки". Расчет ведут по формуле (68).
Проверка восстановительной способности кадмиевой колонки. В стакан или колбу вместимостью 50 см3 вносят пипеткой 20 см3 рабочего раствора нитрата (с. 124) концентрацией 10 мкг NO-3 в см3 и 5 см3 аммиачного буфера и перемешивают.
Через предварительно регенерированную кадмиевую колонку пропускают со скоростью 5 см3/мин сначала 25 см3 приготовленного раствора нитрата калия, когда резервуар над колонкой опустеет, его промывают два раза, используя каждый раз по 15 см3 воды.
После того, как последняя порция воды стечет в колонку, полностью заполняют резервуар водой. Элюат собирают в мерную колбу вместимостью 100 см3 и доводят объем до метки.
45 см3 полученного элюата помещают в мерную колбу вместимостью 100 см3 и проводят цветную реакцию (с. 126). Величину оптической плотности растворов определяют по отношению к холостому раствору.
Если концентрация нитрита в элюате по градировочному графику на нитриты (с. 129) будет ниже 0,70 мкг нитрита в 1 см3 колориметрируемого раствора (т.е. 95 % теоретического значения), то редукционную колонку нельзя использовать для анализа.
В этом случае кадмий необходимо перенести в химический стакан и залить на ночь раствором соляной кислоты, промыть водой и подготовить колонку (с. 124).
Расчет содержания нитратов по варианту II. Если восстановительная способность кадмиевой колонки ≥ 95 %, то количество нитратов в образце (Х), выраженное в мг/кг продукта (в перерасчете на нитрат-ион), может быть рассчитано по формуле
где:
1,35 - коэффициент пересчета нитритов в нитраты;
С - концентрация нитритов, мкг/см3, колориметрируемого раствора, рассчитанная по градуировочному графику, полученному при калибровке стандартных растворов;
m - масса навески продукта, г;
V1 - общий объем экстракта, см3;
V2 - объем фильтрата, взятый на колонку, см3;
V3 - общий объем элюата, см3;
V4 - объем элюата, взятый на цветную реакцию, см3;
V5 - общий объем колориметрируемого раствора, см3.
Регенерация кадмия. По окончании проведения анализа необходимо промыть колонку с кадмием 25 см3 0,1 моль/дм3 (0,1 н) раствора соляной кислоты, 50 см3 дистиллированной воды, 25 см3 разбавленного (1:9) аммиачного буфера (с. 123) и вновь 50 см3 дистиллированной воды.
В случае появления пузырьков газа в колонке, снижения скорости элюации вследствие уплотнения кадмия или длительного перерыва в работе следует перенести кадмий из колонки в химический стакан и залить 100 - 200 см3 0,1 моль/дм3 (0,1 н) раствора соляной кислоты. Перед использованием кадмий декантацией промывают 0,1 моль/дм3 (0,1 н) раствора соляной кислоты 3 - 4 раза по 200 см3, затем несколько раз дистиллированной водой до нейтральной реакции по универсальной индикаторной бумаге. При промывании следует отделять декантацией мелкие частицы кадмия, чтобы избежать в процессе работы чрезмерного уплотнения колонки и снижения скорости элюации.
Примечание: отделенные мелкие частицы кадмия следует хранить в вытяжном шкафу в склянке с притертой пробкой под слоем воды.
2.11. Определение содержания яиц
2.11.1. Качественная реакция
Метод основан на цветной реакции креатинина желтка яиц, который в щелочной среде с насыщенным раствором пикриновой кислоты дает оранжево-красное окрашивание.
Метод применяется для определения наличия яиц в полуфабрикатах и изделиях, указанных в прил. 1, 2.
Метод неприменим для исследования изделий, в состав которых входит мясо, мясной сок или бульон, так как содержат креатинин.
Аппаратура, материалы, реактивы. Весы лабораторные; ступка фарфоровая диаметром 50 - 70 мм; чашка фарфоровая диаметром 50 - 70 мм; пипетка вместимостью 5 см3, градуированная; цилиндр измерительный вместимостью 10 см3; гидроксид натрия, раствор с массовой долей 10 - 15 %; кислота пикриновая, насыщенный раствор (2 г пикриновой кислоты растворяют в 100 см3 дистиллированной воды); вода дистиллированная.
Проведение испытания. Из подготовленной пробы взвешивают 5 - 10 г образца с точностью до 0,01 г, переносят в фарфоровую ступку, растирают с небольшим количеством воды, после чего заливают 5 - 10-кратным количеством воды и настаивают 20 - 25 мин. Содержимое ступки помешивают в течение первых 10 мин каждые 2 - 3 мин, а затем из отстоявшейся вытяжки сливают в фарфоровую чашку 5 - 10 см3 раствора, добавляют 2 - 3 см насыщенного раствора пикриновой кислоты, 5 - 6 капель раствора гидроксида натрия с массовой долей 10 - 15 % и оставляют на 5 мин.
При наличии яиц вытяжка окрашивается в оранжево-красный цвет. При стоянии окраска становится более интенсивной.
2.11.2. Колориметрический метод определения стеролов (по Либерману-Бурхарду)
Метод основан на взаимодействии хлороформного раствора холестерола с уксусным ангидридом и серной кислотой, вследствие чего появляется сине-зеленое окрашивание (реакция Либермана-Бурхарда). Помимо холестерола, реакцию Либермана-Бурхарда дают многие стеролы, в том числе эргостерол и ситостерол. Ненасыщенные стеролы реагируют быстрее насыщенных.
Окрашенные продукты определяют фотометрически.
Аппаратура, материалы, реактивы. Фотоэлектроколориметр или спектрофотометр СФ-4А; штатив для пробирок; весы лабораторные; баня водяная; термометр на 100 °C; чашки фарфоровые диаметром 50 - 70 мм; мерная колба 50 см3 (с притертой пробкой); пробирки емкостью 10 см3 с пробками; колба коническая вместимостью 100 см3 (с притертой пробкой); воронки диаметром 5 - 10 см; хлороформ; уксусный ангидрид; серная кислота концентрированная; сульфат натрия безводный; прокаленный песок; ацетон.
Проведение испытания. Навеску средней пробы изделия (10 г) растирают в фарфоровой чашке с прокаленным песком и подсушивают в сушильном шкафу при 100 - 105 °C в течение 20 - 25 мин. Высушенную пробу растирают пестиком и количественно переносят 20 см3 хлороформа в коническую емкость 100 см3 для экстракции. В изделиях с малой влажностью навеску, растертую с песком, сразу переводят в коническую колбу, куда добавляют 1 г безводного сульфата натрия и 20 см3 хлороформа. Колбу закрывают пробкой с термометром и экстрагируют стеролы в течение 5 мин на водяной бане при 60 - 65 °C. Вытяжку фильтруют через складчатый фильтр в мерную колбу вместимостью 50 см3. Экстракцию повторяют трижды, беря каждый раз по 10 см3 хлороформа. Общий объем хлороформного экстракта должен составлять 50 см3.
Экстракцию стеролов из теста проводят следующим образом: навеску 15 - 17 г растворяют в фарфоровой чашке с песком и сушат в сушильном шкафу при температуре 105 °C. Высушенную пробу растирают до состояния муки и экстрагируют ацетоном. Из экстракта отгоняют растворитель, осадок подсушивают и растворяют в хлороформе, доводя объем последнего до 100 см3.
В три пробирки с притертыми пробками отбирают по 2,5 см3 экстракта, добавляют по 1 см3 уксусного ангидрида и нагревают на водяной бане при температуре 55 - 60 °C 10 мин. Затем к смеси добавляют по 4 капли концентрированной серной кислоты, нагревают 15 мин при температуре 32 - 35 °C и определяют оптическую плотность исследуемого раствора против хлороформа в фотоэлектроколориметре или в спектрофотометре при длине волны 630 нм (кювета 5 мм).
Количество стеролов в исследуемом изделии определяют по формуле
|
|
(69) |
где:
Х - количество стеролов в изделии, мг;
a - количество холестерола, найденное по калибровочной кривой, мг;
в - масса изделия, г;
10 - масса навески, г;
50 - объем хлороформной вытяжки, см3;
2,5 - объем хлороформной вытяжки, взятый для определения, см3.
Калибровочную кривую строят по чистому кристаллическому холестеролу, для чего 10 мг его растворяют в перегнанном хлороформе в мерной колбе вместимостью 100 см3. Из основного раствора отбирают в пробирки микропипеткой по 0,1; 0,3 и 0,5 см3 и добавляют к ним соответственно по 2,4; 2,2 и 2,0 см3 хлороформа. С каждым раствором проводят цветную реакцию и замеряют оптическую плотность приготовленных растворов.
Калибровочную кривую строят в координатах: оптическая плотность - содержание холестерола (мг).
Количество яиц, вложенных в изделие, рассчитывают исходя из того, что в среднем в одном яйце содержится 240 мг холестерола (реакцию Либермана-Бурхарда дают как свободные, так и связанные стеролы), тогда количество вложенных яиц (У) можно рассчитать по формуле
|
|
(70) |
где:
А - найденное количество стеролов, мг;
В - количество стеролов в сырьевом наборе (кроме яиц), мг.
Величину В находят расчетным путем или экспериментально. Среднее содержание стеролов (свободных и связанных) в муке принимают равным 380 мг/кг.
Раздел 3. Контроль качества полуфабрикатов
3.1. Отбор проб полуфабрикатов, подготовка их к анализу
Оценку качества полуфабрикатов начинают с внешнего осмотра тары (ящиков, контейнеров, лотков, функциональных емкостей). Тара должна быть целой, закрытой крышками. Затем просчитывают количество единиц упаковки и взвешивают их для определения массы полуфабрикатов брутто. Для оценки качества полуфабрикатов составляют выемку, вскрывая определенное количество единиц транспортной упаковки. Из вскрытых единиц упаковки для составления средней пробы отбирают определенное количество полуфабрикатов, указанное в действующей нормативно-технической документации (ГОСТ, РСТ и др.).
Отобранные полуфабрикаты оценивают по органолептическим и физико-химическим показателям, определяют массу.
В прил. 1 приведен порядок отбора, подготовки проб к испытаниям и исследуемые физико-химические показатели качества полуфабрикатов.
3.2. Мясные полуфабрикаты
3.2.1. Влажность
Определение ведут, как описано на с. 11.
3.2.2. Качественное определение наполнителя в мясных натуральных рубленых изделиях (бифштексах, шницелях и т.д.)
Метод основан на взаимодействии раствора Люголя (раствор йода в йодиде калия) с крахмалом наполнителей (картофеля, хлеба, каш), в результате чего образуется характерное для каждого наполнителя окрашивание.
Аппаратура, материалы, реактивы. Весы лабораторные; плитка электрическая; колба мерная вместимостью 100, 250 см3; колба коническая вместимостью 100 см3; пипетки вместимостью 1 и 10 см3; вода дистиллированная; раствор Люголя.
Проведение испытания. От пробы, приготовленной, как указано в прил. 1, берут навеску 5 г и помещают ее в коническую колбу, доливают 100 см3 дистиллированной воды, доводят до кипения и отстаивают. 1 см3 отстоявшейся вытяжки помещают в пробирку, разбавляют 10-кратным количеством воды и добавляют 2 - 3 капли раствора Люголя.
При наличии в изделии хлеба вытяжка приобретает интенсивно-синий цвет, переходящий при избытке раствора Люголя в зеленый; картофеля - в лиловый; каши - в синеватый, переходящий при избытке раствора Люголя в грязноватый зеленовато-желтый цвет.
3.2.3. Определение содержания мяса в рубленых полуфабрикатах
Метод основан на том, что экстрактивные вещества мяса с диазотированным белым стрептоцидом (или сульфаниловой кислотой) дают красное окрашивание, интенсивность которого зависит от содержания мяса. Другие компоненты полуфабрикатов и готовых изделий (хлеб, сухари, соль, перец, лук, рис, жир) такой окраски не образуют.
Аппаратура, материалы, реактивы. Фотоэлектроколориметр ФЭК-М (или ФЭК-56, ФЭК-56М, ФЭК-60, КФК); весы лабораторные; баня водяная электрическая; мясорубка; чашки фарфоровые диаметром 10 - 11 см; пипетки вместимостью 1 см3, 5 см3 с делениями; цилиндры вместимостью 250 см3, 100 см3, 50 см3, 10 см3; воронки стеклянные диаметром 5 - 7 см; колбы конические вместимостью 250 см3 с притертыми или резиновыми пробками; пробирки химические; бумага фильтровальная; карбонат Na или кристаллогидрат NaCO3∙10H2O, раствор с массовой долей 15 % в пересчете на безводную соль; стрептоцид белый фармакопейный (аптечный) в порошке (но не из таблеток), раствор с массовой долей 0,5 % в 5 % серной кислоте; серная кислота, раствор с массовой долей 5 %; нитрит натрия (натрий азотистокислый), раствор с массовой долей 0,5 %; спирт этиловый; вода дистиллированная.
Подготовка к испытанию. Раствор белого стрептоцида: 0,5 г белого стрептоцида растворяют в 99,5 см3 раствора серной кислоты с массовой долей 5 %. Раствор хранят в плотно укупоренной склянке из оранжевого стекла (вместо белого стрептоцида можно использовать сульфаниловую кислоту).
Для приготовления раствора нитрита натрия 0,5 г нитрита натрия растворяют в 99,5 см3 дистиллированной воды, фильтруют. Раствор хранят не более 10 суток в плотно укупоренной склянке из оранжевого стекла.
Раствор диазотированного белого стрептоцида свежеприготовленного готовят в день проведения анализа: смешивают 1 объем раствора белого стрептоцида с массовой долей 0,5 % в растворе серной кислоты с 2 объемами раствора нитрита натрия с массовой долей 0,5 %, тщательно перемешивают. Реактив готов к применению через 2 - 3 мин и годен в течение дня.
Спирт этиловый, раствор 30-процентный: готовят смешиванием 30 см3 этилового спирта с 70 см3 дистиллированной воды.
Проведение испытания. Испытание проводится в сравнении с контрольным образцом полуфабриката или готового изделия. Контрольный образец готовят по рецептуре*(15) из того же мяса и других компонентов, используемых для исследуемого (доставленного на анализ) полуфабриката. Контрольный и доставленный образцы исследуют параллельно, как описано ниже.
_______________
*(15) Способ распространяется на следующие рецептуры Сборника рецептур блюд и кулинарных изделий для предприятий общественного питания, 1981 г.: №№ 654-656, 658-660, 662, 668-671, 675-679.
Образец полуфабриката предварительно прогревают (кулинарные изделия не нагревают), затем доводят до комнатной температуры и дважды пропускают через мясорубку. Отбирают пробу массой 10,0 г, помещают ее в сухую фарфоровую чашку диаметром 10 - 11 см, слегка прижимают пестиком для образования лепешки диаметром 6 - 7 см (для равномерного прогрева) и помещают чашку в кипящую водяную баню точно на 20 мин, затем охлаждают на воздухе 5 мин.
Отмеривают цилиндром 200 см3 дистиллированной воды, небольшое количество (около 20 см3) приливают в чашку с навеской, тщательно растирают пестиком до кашицеобразного состояния и с помощью оставшейся в цилиндре воды количественно переносят пробу через воронку в сухую коническую колбу на 250 см3. Колбу закрывают притертой или резиновой пробкой и очень энергично встряхивают вручную 3 - 5 мин, затем оставляют для отстаивания на 15 - 20 мин. Верхний слой (не взмучивая) фильтруют в сухую пробирку через двойной сухой фильтр из медленно фильтрующей бумаги "синяя лента".
Пипеткой переносят 1 см3 фильтрата в другую сухую пробирку и приливают последовательно пипетками 1 см3 раствора карбоната натрия, перемешивают, 1 см3 свежеприготовленного диазотированного белого стрептоцида, перемешивают, затем точно через 1 мин приливают пипеткой 5 см3 этилового спирта и перемешивают.
Через 2 мин после добавления спирта измеряют оптическую плотность образовавшегося красного раствора на ФЭК при зеленом светофильтре (ФЭК-56, ФЭК-56М, КФК - светофильтр № 5; ФЭК-60 - светофильтр № 4) в кювете на 5 мм. В кюветы сравнения помещают дистиллированную воду. Строго соблюдают требования инструкции по работе на ФЭКе, особое внимание следует обращать на чистоту кювет, прозрачность их рабочих граней и измеряемых растворов.
Пример расчета. На испытание поступили полуфабрикаты, приготовленные по 1 колонке рецептуры № 658 Сборника рецептур блюд и кулинарных изделий, 1981 г. По той же рецептуре, из того же вида мяса (говядина) готовят контрольный (стандартный) полуфабрикат с содержанием по норме (в одной порции):
Говядина - 74,00 г (т.е. в формуле расчета С = 74,00);
Хлеб пшеничный - 18,00 г;
Вода - 24 см3;
Сухари - 10 г.
Далее доставленные на испытание и приготовленные контрольные образцы анализировались параллельно.
Результаты измерения оптической плотности Дx = 0,750; Дк = 0,818.
Содержание мяса в порции исследуемого полуфабриката равно:
![]()
Заключение. При закладке по норме 74,00 г мяса максимальные 10 %-ные отклонения (потери при приготовлении, порционировании, погрешности испытания) составляют ±7,40 г, т.е. минимальное содержание мяса в порции равно 74,00 - 7,40 = 66,60 г. Таким образом, исследуемый полуфабрикат соответствует рецептуре.
Содержание мяса в порции (Х, г) рубленых полуфабрикатов определяют по формуле
где:
Дх - оптическая плотность исследуемого образца;
Дк - оптическая плотность контрольного образца;
С - масса мяса (г) в порции контрольного образца.
3.2.4. Определение массовой доли хлеба
В полуфабрикатах из рубленого мяса количество хлеба определяют по крахмалу (с. 74) йодометрическим и ускоренным йодометрическим методом.
3.2.4.1. Определение массовой доли хлеба ускоренным йодометрическим методом (колориметрическим)
Метод основан на измерении интенсивности окраски раствора, полученного при окислении сахаров щелочным раствором сульфата меди II (смесью растворов Фелинга 1 и 2).
Подготовка к испытанию. Раствор гидролизата готовят, как описано выше; растворы Фелинга 1 и 2 (с. 47).
Проведение испытания. 25 см3 гидролизата (при контрольном определении 25 см3 дистиллированной воды) вносят в мерные колбы вместимостью по 100 см3, куда предварительно внесено пипеткой 30 см3 смеси растворов Фелинга 1 и 2, перемешивают и кипятят 2 мин. Затем колбу охлаждают в холодной воде, доводят объем жидкости до метки дистиллированной водой, перемешивают и дают осесть осадку оксида меди (I). Полученный после окисления окрашенный раствор в мерной колбе вместимостью 100 см3 или фильтруют через стеклянный фильтр № 4 для удаления осадка оксида меди (I), или после непродолжительного отстаивания осторожно наливают в кювету толщиной 5 мм фотоэлектроколориметра. Интенсивность окраски измеряют при длине волны 630 нм против дистиллированной воды. По оптической плотности на градуировочном графике находят концентрацию сахара.
Построение градуировочного графика. Глюкозу доводят до постоянной массы в сушильном шкафу при 70 °C, взвешивают 1 г на аналитических весах с точностью до 0,001 г и переносят количественно дистиллированной водой в мерную колбу вместимостью 100 см3, доводят до метки и тщательно перемешивают (основной раствор).
В три мерные колбы вместимостью по 100 см3 пипеткой вместимостью 10 см3 переносят соответственно 10, 20 и 30 см3 основного раствора глюкозы, доводят каждую колбу до метки и перемешивают. Таким образом получают растворы глюкозы с массовой долей соответственно 0,1; 0,2; 0,3 %.
Для проведения реакции в три мерные колбы вместимостью по 100 см3 вносят пипеткой по 30 см3 смеси растворов Фелинга 1 и 2 и по 25 см3 раствора глюкозы с массовой долей 0,1 %, перемешивают и кипятят на плитке 2 мин (считая от начала появления пузырьков). После кипячения колбы тотчас же охлаждают, доводят до метки и после непродолжительного отстаивания для осаждения осадка оксида меди (I) раствор колориметрируют. Таким же образом проводят реакцию с растворами глюкозы массовой долей 0,2 и 0,3 %, а также с дистиллированной водой.
По полученным данным для каждого раствора рассчитывают среднее арифметическое значение оптической плотности и по ним строят градуировочный график (рис. 3).
На оси абсцисс откладывают концентрацию глюкозы (С, г/100 см3), на оси ординат - оптическую плотность (Д).
Обработка результатов испытания. Массовую долю хлеба (Х, %) вычисляют по формуле
|
|
(71) |
где:
a - массовая доля сахара, найденная по градуировочному графику в 100 см3 раствора, г;
m - масса навески продукта, г;
2,5 - коэффициент, учитывающий общий объем раствора;
0,9 - коэффициент пересчета глюкозы на крахмал;
48 - коэффициент пересчета крахмала на хлеб (учитывая массовую долю углеводов в 100 г хлеба).
Рис. 3. Градуировочный график определения сахара по глюкозе
Результаты испытаний вычисляют с точностью до 0,1 %.
За результат испытания принимают среднее арифметическое результатов двух параллельных определений, допустимое расхождение между которыми не должно превышать 0,5 %.
Пример расчета 1. На исследование поступили котлеты "Домашние" средней массой 50 г. Для анализа взяли две навески по 5 г и перенесли их в мерные колбы вместимостью 250 см3. При определении общего количества сахаров после гидролиза крахмала на контрольное титрование 10 см3 раствора гексацианоферрата (II) калия с массовой долей 1 % израсходовано 5,3 и 5,4 см3 гидролизата, в среднем 5,35 см3. Поправка на титр раствора гексацианоферрата (II) калия равна 1,023.
Массовая доля редуцирующих сахаров в изделии составит 9,7 %:
![]()
![]()
Заключение. Вложение хлеба выше нормы на 0,2 % за счет недовложения мяса. При выходе полуфабриката 50 г недовложение мяса составляет 0,1 г или 0,3 % к массе мяса.
Пример расчета 2. В лабораторию доставлены тефтели-полуфабрикат, приготовленные по рецептуре (нетто, г): говядина - 76, хлеб пшеничный (булки городские) - 16, вода - 24, лук репчатый пассерованный - 12, мука пшеничная 1-го сорта - 8; масса полуфабриката - 135 г.
Определено: фактическая масса двух порций полуфабриката (6 шт.) 272 г, что соответствует нормативному выходу.
При определении редуцирующих сахаров, введенных с луком (после гидролиза сахарозы), на титрование 10 см3 гексацианоферрата (II) калия пошло 8,95 и 9,05 см3 раствора сахаров, среднее значение - 9,0, а при исследовании раствора после гидролиза крахмала - 4,1 и 4,2 см3, в среднем - 4,15 см3. Поправка на титр раствора гексацианоферрата (II) калия с массовой долей 1 % составила 1,11.
Массовая доля редуцирующих сахаров, введенных с луком, 2,5 %
![]()
Массовая доля редуцирующих сахаров после гидролиза крахмала 13,5 %:
![]()
Содержание моно- и дисахаридов в городских булках 3,4 %; в 16 г хлеба - 0,56 г, что составляет 0,41 % к массе полуфабриката.
Масса хлеба в одной порции полуфабриката 28,8 г
![]()
Для приготовления тефтелей, кроме хлеба, использовали муку
пшеничную 1-го сорта, содержащую 67,1 % крахмала; 8 г муки соответствует 11,2 г
хлеба ![]() . Общее количество крахмалосодержащих
продуктов в пересчете на хлеб в изделии 27,2 г (16 + 11,2).
. Общее количество крахмалосодержащих
продуктов в пересчете на хлеб в изделии 27,2 г (16 + 11,2).
Заключение. В тефтели вложено на 1,6 г больше крахмалосодержащих продуктов.
3.2.5. Массовая доля соли
Определение соли проводят, как указано на с. 89 (ГОСТ 9957-73).
3.3. Овощи, фаршированные мясным фаршем (голубцы, перец, кабачки, баклажаны)
Содержание фарша. Для определения содержания фарша и оболочки в полуфабрикатах - голубцы, кабачки, перец фаршированные - их взвешивают: кабачков - 4 шт. (2 середины и 2 края), остальных полуфабрикатов - по 3 шт., отделяют фарш, взвешивают его и рассчитывают содержание фарша.
В фарше определяют массовую долю сухих веществ, жира, поваренной соли и риса. При подготовке к испытаниям фарш пропускают через мясорубку, растирают в ступке до однородной консистенции или измельчают в размельчителе тканей с добавлением равного количества воды (60 - 70 °C). Добавленную воду учитывают при расчете.
Массовую долю сухих веществ определяют, как описано на с. 11, жира - на с. 21, количество риса на с. 76.
3.4. Полуфабрикаты из мяса птицы
Исследования проводят так же, как и мясных полуфабрикатов (см. с. 136).
3.5. Рыбные полуфабрикаты
В рыбе специальной разделки незамороженной определяют содержание поваренной соли (см. с. 89), если рыба подвергалась фиксации.
При сомнении в доброкачественности полуфабрикаты направляют для лабораторного исследования с целью определения содержания аммиака, летучих азотистых оснований и сероводорода. Для проведения исследования берут мышечную ткань без кожи и костей и пропускают дважды через ручную мясорубку или один раз через электрическую.
Рубленые полуфабрикаты (биточки, тефтели и др.) контролируют по следующим показателям: массовая доля сухих веществ (см. с. 11), поваренной соли (см. с. 89), хлеба и лука (см. с. 75).
3.6. Блинчики с фаршем
Содержание фарша. Из 5 шт. взвешенных с точностью до 0,1 г блинчиков отделяют фарш от оболочек, не захватывая их. Оболочки взвешивают. По разности между массой блинчиков и оболочек рассчитывают массовую долю фарша в процентах к массе блинчиков.
Массовая доля сухих веществ. Определение массовой доли сухих веществ в фарше описано в табл. 2 (с. 12).
3.7. Овощные полуфабрикаты
3.7.1. Определение содержания остаточного сернистого ангидрида в картофеле сыром очищенном сульфитированном (полуфабрикате) йодометрическим методом
Принцип йодометрического метода состоит в том, что в результате реакции между бисульфатом натрия, гидроксидом натрия и серной кислотой образуется очень непрочная сернистая кислота, которая распадается на воду и сернистый ангидрид. Последний количественно окисляется йодом в серную кислоту.
Аппаратура, материалы, реактивы. Весы лабораторные; ступки; пестики; чашки фарфоровые; колбы конические вместимостью 100, 250 см3; колбы мерные вместимостью 100 и 250 см3; микробюретки вместимостью 2 см3 с ценой деления не более 0,01 см3; цилиндры мерные вместимостью 25 и 50 см3; палочки стеклянные; воронки стеклянные; пипетки вместимостью 1 и 5 см3, градуированные; пипетки вместимостью 10 и 25 см3; пробки резиновые; гидроксид натрия, раствор 1 моль/дм3 (1 н); кислота серная, раствор концентрации C(1/2H2SO4) = 1 моль/дм3 (1 н); кислота соляная плотностью 1,19 г/см3; бихромат калия; йодид калия, раствор массовой долей 10 %; вода дистиллированная; крахмал растворимый, раствор массовой доли 1 %.
Подготовка к испытанию. Раствор бихромата калия: 1,96 г бихромата калия растворяют в воде и доводят объем раствора в мерной колбе до 100 см3. Раствор йода концентрацией 0,005 моль/дм3 (0,01 н): в мерную колбу вместимостью 100 см3 последовательно вносят 25 см3 раствора бихромата калия, 2,5 см3 раствора йодида калия, 5 см3 соляной кислоты, доливают водой до метки и хорошо перемешивают. Тщательно приготовленный раствор не нуждается в проверке титра.
Раствор готовят в количестве дневной потребности в нем.
Проведение испытания. Из подготовленных проб (сульфитированного картофеля) отвешивают с точностью до 0,01 г в фарфоровые чашки две навески по 5 г и смывают их 50 см3 дистиллированной воды в конические колбы вместимостью 100 - 250 см3.
В колбы добавляют по 5 см3 раствора гидроксида натрия, закрывают их пробками, взбалтывают содержимое и оставляют стоять на 15 минут.
Затем в колбы прибавляют по 10 см3 раствора серной кислоты, перемешивают их содержимое, вносят в них по 1 см3 раствора крахмала и тотчас же титруют при взбалтывании раствором йода до появления синей окраски, не исчезающей в течение 2 - 3 секунд.
Одновременно аналогичным способом проводят испытания клубней несульфитированного картофеля, отобранного одновременно с пробами полуфабриката, и сравнивают результаты испытания.
Обработка результатов. Массовую долю сернистого ангидрида в процентах (Х) рассчитывают по формуле
|
|
(72) |
где:
V - объем раствора йода, израсходованный на титрование навески сульфитированного картофеля, см3;
V1 - объем раствора йода, израсходованный на титрование навески несульфитированного картофеля, см3;
0,00032 - количество граммов сернистого ангидрида, окисляющегося 1 см3 раствора йода;
m - масса навески картофеля, г.
За окончательный результат анализа принимают среднее арифметическое результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 0,001 %.
Массовая доля сернистого ангидрида в сульфитированном картофеле не должна превышать допустимой нормы 0,002 % к массе.
При получении результатов испытаний выше допустимой нормы сырой очищенный сульфитированный картофель повторно промывают водой, снова проверяют остаточное содержание в нем сернистого ангидрида и, при соответствии норме, допускают к реализации.
При получении неудовлетворительных результатов при повторном испытании вся партия бракуется и сдаче-приемке не подлежит. Этот результат является окончательным.
3.8. Голубцы (полуфабрикат)
Содержание фарша. Для определения соотношения массы капусты и фарша взвешивают 3 полуфабриката. Полуфабрикат развертывают, отделяют фарш, взвешивают его и рассчитывают соотношение капусты и фарша. Фарш измельчают в ступке или в измельчителе тканей.
В подготовленном к испытанию фарше определяют массовую долю сухих веществ, жира методом Гербера (с. 31), кислотность (с. 78) и поваренную соль (с. 89).
3.9. Биточки (котлеты) крупяные
3.9.1. Массовая доля сухих веществ
Массовую долю сухих веществ определяют, как указано на с. 11.
3.9.2. Массовая доля сахара
Аппаратура, материалы, реактивы. Водяная баня; термометр ртутный стеклянный лабораторный на 100 - 150 °C; колбы мерные вместимостью 100, 250 см3; стаканы химические вместимостью 50 см3; фарфоровая ступка с пестиком; пипетки вместимостью 5, 10, 50 см3; воронки стеклянные; палочки стеклянные; сульфат цинка, раствор с массовой долей 4,5 %; гидроксид натрия, растворы с массовой долей 4 % и 20 %; соляная кислота плотностью 1,19 г/см3; метиловый оранжевый, раствор с массовой долей 0,1 %.
Проведение испытания. Содержание сахара в растворе, который будет получен из навески, должно быть в пределах 0,4 - 0,8 %. Навеску массой 5 - 10 г количественно переносят в мерную колбу вместимостью 250 см3, смывая частицы в колбу дистиллированной водой примерно до половины объема колбы. При частом взбалтывании содержимое колбы оставляют на 1 ч для извлечения сахаров в раствор. Мерным цилиндром добавляют 20 см3 раствора сульфата цинка и 5 см3 раствора гидроксида натрия с массовой долей 4 %. Содержимое колбы доводят до метки дистиллированной водой, перемешивают и фильтруют в сухую колбу. В фильтрате определяют сахар до инверсии перманганатным методом.
Для определения сахаров после инверсии сахарозы 50 см3 фильтрата пипеткой переносят в мерную колбу вместимостью 100 см3, приливают цилиндром 5 см3 соляной кислоты, помещают в колбу термометр и ставят ее в водяную баню, нагретую до температуры 80 °C, доводят температуру раствора в течение 2 - 3 мин до 67 - 70 °C и при этой температуре выдерживают раствор точно 8 мин. Затем, охладив раствор до 20 °C, удаляют термометр, предварительно ополоснув его дистиллированной водой, нейтрализуют соляную кислоту раствором гидроксида натрия с массовой долей 20 % до получения желтого окрашивания по метиловому оранжевому, доводят объем до метки дистиллированной водой, перемешивают. В полученном растворе определяют сахара перманганатным способом.
Массовую долю сахаров до и после инверсии сахарозы в граммах рассчитывают по формулам (19), (20), где Р = 100, а массовую долю сахарозы в процентах - по формуле (21).
3.10. Полуфабрикаты (тесто охлажденное)
3.10.1. Влажность
Определение проводят, высушивая навеску в сушильном шкафу или в аппарате ВЧ (с. 12).
3.10.2. Массовая доля жира
Жир в тесте определяют экстракционно-весовым методом с предварительным гидролизом крахмала (с. 29).
3.10.3. Массовая доля сахара
В полуфабрикатах (тесто) определяют редуцирующие сахара после инверсии сахарозы и выражают их содержание в сахарозе на сухое вещество (ГОСТ 5672-68).
Аппаратура, материалы, реактивы. Используемые аппаратура и материалы указаны выше. Реактивы: сульфат цинка 7-водный, раствор массовой концентрацией 150 г/дм3; гидроксид натрия, растворы с массовой концентрацией 40 г/дм3 и 100 г/дм3; метиловый красный, водно-спиртовой раствор с массовой долей 0,2 %; соляная кислота, раствор с массовой долей 20 %.
Проведение испытания. Навеску теста (30 г дрожжевого, 12,5 г дрожжевого слоеного, 7 г песочного), взвешенную с точностью до 0,01 г, растирают с помощью 100 - 120 см3 дистиллированной воды, прибавляя ее порциями, и количественно переносят в мерную колбу вместимостью 250 см3. Колбу с содержимым нагревают на водяной бане при температуре не выше 60 °C в течение 15 мин при частом взбалтывании, затем содержимое колбы охлаждают до комнатной температуры. К охлажденному раствору прибавляют 10 см3 раствора сульфата цинка и 10 см3 раствора гидроксида натрия или 40 г/дм3 гидроксида калия (концентрацией 56 г/дм3). Содержимое колбы взбалтывают, доводят дистиллированной водой до метки, перемешивают и фильтруют через складчатый фильтр в сухую колбу.
Для гидролиза сахарозы 50 см3 полученного фильтрата пипеткой переносят в мерную колбу вместимостью 100 см3, добавляют 5 см3 раствора соляной кислоты массовой долей 20 %. Колбу погружают в водяную баню, нагретую до 70 °C, и выдерживают 8 мин при этой температуре. Содержимое колбы быстро охлаждают под струей воды до температуры 20 °C, нейтрализуют раствором гидроксида натрия концентрацией 100 г/дм3 или безводными углекислыми солями натрия по метиловому красному до появления желто-розового окрашивания. Колбу доливают водой до метки. Содержимое колбы перемешивают и определяют редуцирующие сахара перманганатным методом по Бертрану (с. 47).
Массовую долю общего сахара в сахарозе на сухое вещество рассчитывают по формуле (18).
3.10.4. Определение общей кислотности, щелочности
Кислотность. Определение проводят в соответствии с ГОСТ 5898-87 и 5670-51 как указано на с. 79.
Щелочность. Этот показатель определяют в полуфабрикатах, приготовленных с химическими разрыхлителями, как описано на с. 81.
3.11. Полуфабрикаты для тортов и пирожных
Отбор проб полуфабрикатов для тортов и пирожных и подготовка их к испытаниям, определяемые физико-химические показатели приведены в прил. 1.
При подготовке проб отделочных полуфабрикатов (крема, начинки, помады, желе, сиропа) берут точечные пробы, соединяют их и перемешивают, масса объединенной пробы не менее 500 г.
Для определения физико-химических показателей полуфабрикатов из разных мест объединенной пробы отбирают пробу для лабораторных испытаний массой не менее 100 г.
Пробы отделочных полуфабрикатов должны храниться при температуре не выше 6 °C.
Анализ выпеченных полуфабрикатов проводят не ранее чем через 16 ч после изготовления.
3.11.1. Влажность
Определение проводят в соответствии с ГОСТ 5900-73 (см. табл. 2).
3.11.2. Массовая доля жира
В полуфабрикатах жир определяют рефрактометрическим методом, экстракционно-весовым с предварительным гидролизом крахмала навески (см. с. 29) в соответствии с ГОСТ 5899-85.
3.11.3. Обнаружение замены сливочного масла маргарином
Для анализа берут 4 - 5 г крема, 30 - 50 г выпеченного полуфабриката (из пробы, измельченной после удаления корочек) и проводят определение, как описано на с. 43.
3.11.4. Массовая доля сахара
При использовании любого из указанных на с. 52, 59, 61 методов, определения находят общий сахар после инверсии сахарозы и выражают его в сахарозе на сухое вещество.
Аппаратура, материалы, реактивы. Баня водяная; бумага фильтровальная; бумага индикаторная универсальная или лакмусовая; весы лабораторные; воронки; капельницы; колбы конические вместимостью 250 см3; колбы мерные вместимостью 100, 200, 250 см3; пипетки мерные вместимостью 5, 10, 25, 50 см3; палочки стеклянные; термометр с диапазоном измерения 0 - 150 °C с ценой деления 1 °C; цилиндры мерные вместимостью 10, 25, 100 см3; гидроксид натрия, растворы концентрации 40 г/дм3, 100 г/см3; сульфат цинка 7-водный, раствор концентрации 0,5 моль/дм3 (1 н); соляная кислота плотностью 1,19 г/см3; метиловый оранжевый, раствор с массовой долей 0,1 %; фенолфталеин, спиртовой раствор с массовой долей 1 %; вода дистиллированная; карбонат натрия 10-водный, раствор с массовой долей 15 %. Проведение испытания. Навеску измельченного полуфабриката взвешивают в стакане вместимостью 50 см3, добавляют в нее дистиллированную воду, нагретую до 60 - 70 °C. Если полуфабрикат растворяется без остатка (сахарные сиропы, желе, помада), то полученный в стакане раствор охлаждают и переносят в мерную колбу вместимостью 250 см3. Остатки навески смывают дистиллированной водой, доливают колбу водой до метки и хорошо перемешивают жидкость.
Если полуфабрикат содержит вещества, нерастворимые в воде (мешающие несахара - белки, жиры, пектины, крахмал и т.д.), то навеску при необходимости растирают в ступке с теплой водой и переносят в мерную колбу вместимостью 250 см3, смывая нерастворимые частицы в колбу дистиллированной водой примерно до 1/2 объема колбы. Органические кислоты, содержащиеся в навеске, нейтрализуют раствором карбоната натрия с массовой долей 15 % до pH = 7,0, применяя для контроля универсальную индикаторную бумагу. Колбу помещают на водяную баню, нагретую до 60 °C; при этой температуре, временами взбалтывая, выдерживают 15 мин. Смесь охлаждают и осаждают несахара, прибавляя к раствору в колбе 10 см3 раствора сульфата цинка, если масса навески менее 5 г, или 15 см3, если масса навески более 5 г и гидроксид натрия концентрацией 40 г/дм3, объем которого устанавливают отдельным опытом при титровании соответствующего объема раствора сульфата цинка с фенолфталеином. Содержимое колбы взбалтывают, доводят дистиллированной водой до метки, перемешивают, оставляют на 10 - 15 мин, затем фильтруют в сухую колбу. Фильтрат должен быть прозрачным.
Для инверсии сахарозы в мерную колбу вместимостью 100 или 200 см3 вносят пипеткой соответственно 50 или 100 см3 полученного фильтрата, затем в колбу на 100 см3 приливают 5 см3 (или 10 см3 в колбу вместимостью 200 см3) концентрированной соляной кислоты, опускают в колбу термометр и ставят ее в водяную баню, нагретую до 80 - 85 °C. Доводят температуру раствора в течение 2 - 3 мин до 67 - 70 °C и при этой температуре выдерживают раствор точно 5 мин. Затем, охладив раствор до температуры 20 °C, удаляют термометр, предварительно ополоснув его водой, нейтрализуют соляную кислоту раствором гидроксида натрия или калия (25 г в 100 см3 воды), к концу нейтрализации приливают гидроксид натрия или калия концентрацией 100 г/дм3 до получения желтого окрашивания по метиловому оранжевому или индикаторной бумаге. Раствор в колбе доводят до метки дистиллированной водой и перемешивают. В полученном растворе определяют общий сахар после гидролиза сахарозы методами, предусмотренными НТД.
3.11.5. Определение содержания этилового спирта для промочки кондитерских полуфабрикатов. Бихроматный метод
Метод основан на окислении спирта, отогнанного из сиропа, бихроматом калия в присутствии серной кислоты до уксусной кислоты. Не израсходованный на окисление бихромат калия определяют йодометрически. Содержание спирта рассчитывают по количеству израсходованного на его окисление бихромата калия.
Аппаратура, материалы, реактивы. Прибор для перегонки спирта; колба мерная вместимостью 50 см3; колбы конические вместимостью 100 см3 и 500 см3 (с притертой пробкой); пипетка вместимостью 10 см3; мерный цилиндр вместимостью 10 см3; капельница; бюретка вместимостью 25 см3; бихромат калия, раствор концентрации 0,032 моль/дм3 (9,4064 г в 1 дм3); кислота серная концентрированная плотностью 1,84 г/см3; тиосульфат натрия, раствор концентрации 0,1 моль/дм3; раствор крахмала с массовой долей 1 % (свежеприготовленный); йодид калия.
Проведение испытания. Навеску исследуемого сиропа массой 6 г берут в стеклянный стакан с точностью до 0,01 г и переносят в перегонную колбу с помощью 75 см3 дистиллированной воды. В приемную мерную колбу вместимостью 50 см3 наливают 5 - 7 см дистиллированной воды и опускают в нее узкий конец холодильника; колбу помещают в смесь воды со льдом. Перегонную колбу соединяют через каплеуловитель с холодильником и отгоняют спирт до тех пор, пока приемная колба не наполнится водным раствором спирта до 4/5 объема. Колбу отставляют, доводят содержимое дистиллированной водой до метки и тщательно перемешивают. Затем в конической колбе вместимостью 100 см3 готовят окислительную смесь, для чего наливают 10 см3 раствора бихромата калия и по стенке 5 см3 концентрированной серной кислоты. К остывшей смеси добавляют по каплям при непрерывном взбалтывании 10 см3 перемешанного полученного дистиллята.
Колбу соединяют с воздушным холодильником, доводят жидкость до кипения и кипятят в течение 10 мин при слабом кипении. Содержимое переносят в коническую колбу вместимостью 500 см3 с помощью 300 см3 дистиллированной воды. В колбу вносят 1 г йодида калия, плотно закрывают пробкой и оставляют на 2 мин в темноте. Выделившийся йод титруют раствором тиосульфата натрия до грязно-зеленого окрашивания, затем добавляют 5 - 6 капель раствора крахмала и дотитровывают до светло-зеленого цвета. Если жидкость стала зеленой или на титрование пошло менее 8 см3 тиосульфата натрия, отгон разбавляют и повторяют определение.
Обработка результатов. Массовую долю спирта (Х, %) рассчитывают по формуле
|
|
(73) |
где:
V1 - объем раствора бихромата калия концентрацией 0,032 моль/дм3, взятый для окисления спирта, см3;
V2 - объем раствора тиосульфата концентрации 0,1 моль/дм3, пошедший на титрование йода, см3;
2 - коэффициент перерасчета раствора бихромата калия концентрации 0,032 моль/дм3 в 0,016 моль/дм3;
V - объем отгона, см3;
0,00115 - объем спирта, окисляемого в 1 см3 0,016 моль/дм3 раствора бихромата калия, г;
m - навеска сиропа, г;
10 - объем отгона для окисления, см3.
Допустимое расхождение между параллельными определениями не должно превышать 0,01 %.
Полученные результаты сравнивают с расчетными по рецептуре. Допустимое отклонение в меньшую сторону от расчетного - не более 0,2 %.
3.11.6. Расчет содержания этилового спирта в сиропе для промочки кондитерских полуфабрикатов
Пример расчета. В сироп для промочки по рец. 56 (95) "Сборника рецептур мучных кондитерских и булочных изделий для общественного питания" (1986 г.) входят виды сырья (нетто, кг), указанные в табл. 30.
Таблица 30
|
Сырье |
Расход сырья массой нетто на 1 т, кг |
Содержание сухих веществ |
Содержание спирта |
|||
|
% |
кг |
в объемных % |
в массовых % |
кг |
||
|
Сахар-песок |
513,07 |
99,85 |
512,3 |
- |
- |
- |
|
Эссенция ромовая |
1,92 |
0,00 |
0,0 |
- |
- |
- |
|
Коньяк или вино десертное |
47,95 |
0,00 |
0,0 |
40 |
33,3* |
15,96 |
|
Масса сырья |
562,94 |
- |
512,3 |
- |
- |
15,96 |
|
Выход |
1000,00 |
50,00 |
500,0 |
- |
- |
15,58 |
|
|
100 |
50,00 |
50,0 |
- |
- |
1,56 |
_____________
* Объемные проценты переводят в массовые проценты, используя табл. 42.
В соответствии с рецептурой содержание спирта в массовых процентах в сиропе для промочки кондитерских полуфабрикатов составит 1,6. Влажность сиропа 50 ± 4 %.
Раздел 4. Контроль качества блюд и кулинарных изделий
4.1. Отбор проб
Отбор проб блюд и кулинарных изделий работник лаборатории проводит на раздаче из порций, подготовленных для отпуска (на предприятиях самообслуживания), или по выполнении заказа (при обслуживании официантами). В прил. 1, 2 приведены количества блюд, изделий, подлежащих выемке для определения средней массы и физико-химического анализа.
Помимо порции первого или сладкого блюда из числа подготовленных к отпуску, отбирают дополнительно из котлов на раздаче по одной порции блюда того же наименования, при отборе молочных супов и горячих напитков с молоком - пробу молока, используемого для их приготовления. Блюда, взятые из котлов, являются контрольными и исследуются отдельно, отбор их должен производиться с особой тщательностью. При отборе проб супов содержимое котла хорошо перемешивают, отливают не менее 5 порций в отдельную кастрюлю, разливают по тарелкам и отбирают одну порцию.
Кулинарные изделия из мяса, птицы, рыбы, кролика, органолептические показатели и масса которых соответствуют норме, на анализ не отбирают. Если масса изделий ниже нормативной, внешний вид их свидетельствует о том, что кулинарная обработка проведена неправильно (пережарены, подсушены) или имеется подозрение на недоброкачественность, на анализ отбирается блюдо целиком, а также дополнительно из котлов на раздаче - по 200 г гарнира и соуса, с которыми отобранное блюдо отпускается. Пробу гарнира берут из центра котла, отступая на 2 - 3 см от стенки, после тщательного перемешивания его содержимого, соус перед отбором пробы тщательно перемешивают шумовкой.
При отборе проб блюд из рубленого мяса, птицы, рыбы, кролика с наполнителем, помимо гарнира и соуса, отбирают дополнительно контрольные основные изделия (котлеты, биточки и др.) или полуфабрикаты для них.
Алкогольные коктейли в количестве двух порций отбирают методом контрольной закупки и параллельно готовят две порции контрольного образца. Контрольное приготовление производится в присутствии лица, отобравшего пробу, из бутылок заводской упаковки в строгом соответствии с рецептурой, с использованием мерной посуды, имеющей клеймо государственной поверки. Исследуемый и контрольный образцы должны быть приготовлены из компонентов одной партии.
Бутылки с напитками, использованными для приготовления исследуемого образца, укупоривают, опечатывают и берут в лабораторию для анализа. Если коктейль приготовлен с использованием импортного алкогольного напитка, для анализа отбирают бутылку заводской упаковки. Другие компоненты (сок, сироп, компот) отбирают в количестве не менее 200 г.
Коктейли с молочными продуктами отбирают для анализа методом контрольной закупки в количестве двух порций (из одного миксера) и параллельно готовят две порции контрольного образца. Две порции исследуемого и контрольного образцов переносят в посуду лаборатории, взвешивают.
Напиток "Кофе черный", изготовляемый в электрокофеварках, отбирают для анализа в количестве одной порции методом контрольной закупки и параллельно готовят порцию контрольного образца из зерен кофе. Определяют объем (массу) контрольного и исследуемого напитков.
Пробы, отобранные для физико-химического анализа, аккуратно, по возможности без потерь, переносят в чистую, сухую, предварительно взвешенную посуду лаборатории. При переносе пробы жидкого блюда тщательно очищают ложкой приставшие к тарелке плотные частицы и присоединяют их к пробе.
При переносе пробы второго мясного или рыбного блюда гарнир и соус счищают с основного изделия и присоединяют их к общей массе гарнира с соусом. Основное изделие взвешивают и оставляют на производстве*(16). Затем в посуду лаборатории переносят в первую очередь часть гарнира с соусом, остальной частью гарнира собирают оставшиеся на тарелки жир и соус и переносят в ту же посуду.
Посуду с пробами закрывают крышками, завертывают в бумагу, обвязываются шпагатом, пломбируют.
______________
*(16) Изделия с заниженной массой отбирают для анализа в лабораторию.
4.2. Холодные блюда
Физико-химические показатели, по которым контролируют соблюдение норм вложения сырья в холодные блюда, и методы анализа приведены в прил. 2.
При подготовке к испытанию овощных салатов и винегретов порцию блюда (салаты из свежих огурцов, помидоров - 2 порции) взвешивают и переносят в размельчитель тканей. Остатки овощей смывают дистиллированной водой (60 - 70 °C), количество которой, а также продолжительность гомогенизации приведены в табл. 31.
Таблица 31
Количество добавляемой воды, продолжительность гомогенизации проб и плотность серной кислоты, используемой для сжигания навески при определении содержания жира методом Гербера
|
Наименование изделия |
Количество воды в см3 на 100 г блюда* |
Время гомогенизации, мин |
Плотность серной кислоты |
|
Салат из свежей капусты |
50 |
5 - 6 |
1,7 |
|
Салат из помидоров и огурцов со сметаной |
25 - 30 |
1 - 2 |
1,68 - 1,7 |
|
Салат из редиса со сметаной |
50 - 70 |
4 - 5 |
1,68 - 1,7 |
|
Салат из свеклы со сметаной |
50 |
2 - 3 |
1,7 |
|
Салат из квашеной капусты |
50 |
5 - 6 |
1,7 |
|
Салат с картофелем, винегреты, салат мясной |
100 |
4 - 5 |
1,65 |
|
Морковь тертая со сметаной |
50 - 70 |
4 - 5 |
1,68 - 1,7 |
______________
* Учитывается количество воды, расходуемое на ополаскивание посуды, в которой доставлена проба, отмывание основного изделия от соуса и добавляемое на измельчение пробы.
При подготовке салатов и винегретов для определения содержания витамина C их измельчают ножом из нержавеющей стали, а затем тщательно растирают в ступке. Если для гомогенизации пробы используют миксер с выносными ножами, то пробу измельчают в посуде, в которую она отобрана.
Мясные и рыбные салаты. Порцию блюда взвешивают. Из мясных салатов кусочки мяса отделяют пинцетом, переносят на мелкое сито, обмывают горячей дистиллированной водой для удаления соуса, дают воде стечь в течение 2 - 3 мин, слегка просушивают фильтровальной бумагой и взвешивают. Промывные воды присоединяют к овощной массе салата. Оставшейся частью воды смывают остатки салата из посуды, присоединяют ее к пробе и измельчают смесь в гомогенизаторе.
При подготовке к анализу рыбных салатов их гомогенизируют целиком.
Рыба под майонезом, маринадом. Порцию блюда взвешивают, осторожно скальпелем счищают соус и переносят его в гомогенизатор. Куски рыбы обмывают отмеренным количеством воды с температурой 70 °C, обсушивают поверхность фильтровальной бумагой и взвешивают. Майонез с овощным гарниром (или маринадом) гомогенизируют, добавив промывные воды.
Холодные блюда из мяса и мясных продуктов. Порцию блюда взвешивают. Куски мяса переносят на сито, обмывают горячей водой, дают воде стечь в течение 2 - 3 мин, слегка обсушивают фильтровальной бумагой и взвешивают. Воду после ополаскивания мяса соединяют с соусом и гарниром и смесь измельчают в размельчителе тканей.
Заливные блюда из рыбы, мяса и мясных продуктов, студни. Взвешивают 2 порции, осторожно на водяной бане расплавляют желе, смывают его остатки теплой водой, поверхность кусков основного продукта обсушивают фильтровальной бумагой и взвешивают.
Студень разогревают до 80 °C, переносят на сито, дают жидкости стечь в течение 10 мин, а затем взвешивают плотную часть.
Масса мяса, птицы, плотной части студня может отличаться на величину, указанную в табл. 1.
4.2.1. Массовые доли сухих веществ и жира
Массовую долю сухих веществ определяют взвешиванием, как указано выше, жира - по методу Гербера (см. с. 31) или экстракционно-весовым методом. Результаты анализов по этим показателям сравнивают с расчетными данными (см. п. 7.3).
4.3. Первые блюда
Физико-химические показатели, по которым контролируют соблюдение норм вложения сырья в первые блюда и методы их анализа, приведены в прил. 2.
Подготовка к испытанию первых блюд сводится к их гомогенизации. Доставленный в лабораторию образец супа разогревают до 75 °C в той посуде, в которой он был доставлен, извлекают и взвешивают находящиеся в супе мясо, птицу, рыбу и сравнивают их массу с выходом по рецептуре. Дальнейшая подготовка первых блюд проводится с выпариванием или без него. При выпаривании порцию супа переносят в выпарительную чашку, смывая остатки дистиллированной водой, и выпаривают более чем на половину объема, избегая подгорания. После упаривания суп взвешивают, переносят в размельчитель тканей и гомогенизируют в течение 1 мин. При определении сухих веществ и жира расчет ведут на массу упаренного блюда.
Пюреобразные супы рекомендуется также обрабатывать в размельчителе тканей для равномерного распределения жира, который обычно собирается на поверхности.
Супы вязкой консистенции (с овсяной и перловой крупами) гомогенизируют без выпаривания.
Прозрачные супы исследуют без предварительной подготовки их, особенно если гарнир к ним подается отдельно. При отпуске гарнира вместе с бульоном (в одной тарелке) последний осторожно отделяют от гарнира, сливая в сухую чистую тарелку.
В порции сборной мясной солянки после подогрева и взвешивания отделяют с помощью пинцета мясные продукты, взвешивают их и сравнивают массу с данными рецептуры, учитывая, что потери при вторичной тепловой обработке для колбасы и сосисок составляют 15 %, для птицы - 15 %, для ветчины - 30 %. Например, в мясную солянку по рецептуре входят: 20 г сосисок, 20 г колбасы, 20 г ветчины. Общая масса продуктов - 60 г. С учетом потерь при вторичной тепловой обработке масса сосисок составит 17 г, колбасы - 17 г, ветчины - 14 г, что в сумме равно 48 г. Следовательно, при анализе должно быть обнаружено не менее 48 г мясных продуктов.
Сладкие супы взвешивают, отделяют плотную часть, взвешивают ее, присоединяют к жидкой части и гомогенизируют.
4.3.1. Массовые доли сухих веществ и жира
Массовую долю сухих веществ определяют высушиванием навески в сушильном шкафу или в аппарате ВЧ (см. с. 8, 15), жира - методами Гербера или весовым с экстракцией жира в микроразмельчителе (см. с. 23, 31). Результаты анализов по этим показателям сравнивают с расчетными данными по рецептуре (теоретическими) или минимальными (см. п. 7.3).
4.3.2. Плотная часть супа
В сладких супах с фруктами определяют массу части в порции, взятой работником лаборатории при отпуске, а также в пяти порциях, отобранных на производстве. По количеству плотной части в одной порции судят о правильности порционирования, а в пяти порциях - о полноте закладки сухофруктов. Для определения плотной части в пяти порциях супа их соединяют вместе, взвешивают, после чего отделяют плотную часть, пользуясь металлическим ситом с жестяным корпусом высотой 10 - 15 см или дуршлагом. Через 10 мин плотную часть супа взвешивают с точностью до 1 г.
Если суп приготовлен с крупяным или другим гарниром, гарнир отделяют от фруктов и также взвешивают.
4.3.3. Массовая доля сахара
Содержание сахара контролируют в сладких супах, определяя редуцирующие сахара до и после инверсии сахарозы.
Аппаратура, материалы, реактивы. Весы лабораторные; баня водяная; термометр лабораторный с диапазоном измерения 0 - 100 °C с ценой деления 1 °C; стаканы химические вместимостью 50 и 100 см3; колбы мерные вместимостью 100, 250 см3; колбы конические вместимостью 100, 250 см3; палочки стеклянные; воронки стеклянные; пипетки вместимостью 5, 10 и 50 см3; гексацианоферрат (II) калия, раствор массовой концентрации 150 г/дм3; сульфат цинка, раствор массовой концентрации 300 г/дм3; соляная кислота плотностью 1,19, раствор с массовой долей 20 %; гидроксид натрия, раствор массовой концентрации 100 г/дм3; метиловый красный, водно-спиртовой раствор массовой концентрации 2 г/дм3.
Подготовка к испытанию. Навеску подготовленного блюда (изделия) взвешивают с точностью до 0,01 г из такого расчета, чтобы в 100 см3 полученного раствора содержалось 0,2 - 0,4 г*(17) редуцирующих сахаров. Массу навески (m, г) вычисляют по формуле
где:
С - оптимальное (в зависимости от метода) содержание редуцирующих сахаров в 100 см3 раствора навески, г;
У - вместимость мерной колбы, см3;
Р - предполагаемая массовая доля редуцирующих сахаров в исследуемом блюде (изделии), %.
_________________
*(17) Содержание редуцирующих сахаров до инверсии в 100 см3 раствора, приготовленного для титрования, должно быть примерно: для перманганатного метода - 0,3 - 0,4 г, йодометрического - 0,4 г, цианидного - 0,2 - 0,4 г; сахара после инверсии - для перманганатного метода - 0,6 - 0,8 г, йодометрического - 0,8 г, цианидного - 0,4 - 0,8 г.
Навеску гомогенизированного супа (15 г) берут в химическом стакане вместимостью 50 - 100 см3 и количественно переносят в мерную колбу вместимостью 250 см3, смывая нерастворимые частицы в колбу теплой дистиллированной водой (50 °C) примерно до половины объема колбы. Колбу встряхивают в течение 3 - 5 мин, затем добавляют по 2 см3 растворов гексацианоферрата (II) калия и сульфата цинка для осаждения несахаров. Содержимое колбы взбалтывают, доводят дистиллированной водой до метки, перемешивают и оставляют на 20 мин для выпадения осадка, а затем фильтруют в сухую колбу. В фильтрате определяют содержание редуцирующих сахаров перманганатным методом по Бертрану (см. с. 47) или цианидным методом (см. с. 55).
Для определения общего сахара проводят инверсию сахарозы: для этого в мерную колбу вместимостью 100 см3 вносят пипеткой 50 см3 полученного фильтрата, 5 см3 раствора соляной кислоты и помещают колбу на 8 мин в водяную баню, нагретую до 70 °C. Затем колбу быстро охлаждают до температуры 20 °C и нейтрализуют соляную кислоту раствором гидроокиси натрия массовой концентрации 100 г/дм3 в присутствии метилового красного до появления желто-розового окрашивания, доводят водой до метки, перемешивают. В полученном растворе определяют общий сахар тем же методом, что и редуцирующие сахара до инверсии.
Массовую долю сахарозы (S, г на порцию) в сладких супах определяют по формуле (21) и сравнивают ее с рецептурой.
4.3.4. Массовая доля молока
Содержание молока в молочных супах определяется по лактозе. Метод основан на том, что в продуктах, которые используют для приготовления супов, содержится незначительное количество редуцирующих сахаров.
Аппаратура, материалы, реактивы. Аппаратура и материалы те же, что указаны на с. 159: сульфат цинка, раствор массовой концентрации 200 г/дм3; гидроксид натрия, раствор концентрации 2,5 моль/дм3.
Подготовка к испытанию. Навеску упаренного и гомогенизированного супа массой 15 г переносят в мерную колбу вместимостью 250 см3, смывая остатки дистиллированной водой. Общее количество воды должно быть не более половины объема колбы. Колбу помещают в водяную баню с температурой 60 °C и выдерживают 15 мин, периодически помешивая, затем охлаждают до температуры 20 °C. Для осаждения несахаров в колбу вносят 3 см3 раствора сульфата цинка и 1,5 см3 раствора гидроксида натрия. Содержимое колбы встряхивают 2 - 3 мин, доводят дистиллированной водой до метки, перемешивают, дают отстояться в течение 3 - 5 мин и фильтруют в сухую колбу. В фильтрате определяют редуцирующие сахара (лактозу) ускоренным цианидным методом.
Одновременно определяют содержание лактозы в молоке, используемом для приготовления супа. Подготовку его к испытанию проводят так же, как и супа.
Количество молока в порции (У, г) вычисляют по формуле
где:
Х - количество лактозы в выпаренной порции супа, г;
А - количество лактозы в молоке, используемом для приготовления супа, %.
Результат испытания сравнивают с количеством молока по рецептуре, с учетом допустимых отклонений ±10 %.
Если содержание лактозы в молоке определить не представляется возможным, то принимают его равным 4,7 % (содержание лактозы в молоке согласно таблицам химического состава пищевых продуктов. М., 1987 г.).
4.4. Вторые блюда
В прил. 2 приведены физико-химические показатели, по которым контролируют соблюдение норм вложения сырья во вторых блюдах.
Подготовка проб к испытанию заключается в следующем: блюда, поступившие на исследование, подогревают до температуры 65 °C, взвешивают (вместо с гарниром и соусом), затем взвешивают отдельно основной продукт (мясо, рыбу, котлеты, сырники, блинчики и др.).
Натуральные куски мяса, рыбы, птицы перед взвешиванием тщательно освобождают от гарнира и соуса.
В изделиях с двойной панировкой (мука, льезон, сухари) определяют количество панировки и выход мяса, рыбы, птицы. Для этого 3 - 5 изделий взвешивают, освобождают их с помощью скальпеля от панировки, снова взвешивают и определяют среднюю массу. Прибавляя к средней массе потери массы при тепловой обработке, находят фактическую массу нетто продукта и сравнивают с массой нетто по рецептуре. Если панировку удалить невозможно (например, мучную панировку с куска жареной рыбы), массу ее принимают равной указанной в Сборнике рецептур блюд, 1981 г.
Если масса мяса, рыбы, птицы, изделий из рубленой и котлетной массы ниже нормы, определяют количество сухих веществ, т.к. уменьшение массы может быть вызвано неправильно проведенной тепловой обработкой (пережариванием, повторным разогревом). Для определения сухих веществ натуральные изделия после удаления костей дважды пропускают через мясорубку, после чего массу растирают в ступке. Изделия из котлетной и рубленой массы (котлеты, биточки, бифштекс рубленый и др.) растирают в ступке или дважды пропускают через мясорубку, а затем перемешивают. Фаршированные изделия (рулеты, зразы) подготавливают так же, удалив предварительно фарш. По рецептуре находят массу полуфабриката (с учетом норм потерь при тепловой обработке), а по таблицам химического состава пищевых продуктов, 1987 г., подсчитывают количество сухих веществ. Последнее сравнивают с фактическим содержанием сухих веществ в изделии и делают заключение о соблюдении рецептуры.
Количество жира, израсходованного на обжарку изделий из мяса, рыбы, птицы, кролика, не учитывают.
В натуральных рубленых изделиях (бифштекс, шницель и др.), помимо определения массы, проводят качественную реакцию с йодом на присутствие крахмала. Панировочные изделия (шницель) предварительно освобождают от корочки. По рецептуре в эти изделия хлеб не входит, поэтому положительная реакция на йод (появление фиолетово-синей окраски) будет свидетельствовать о нарушении рецептуры.
Из блюд, приготовленных с соусом (бефстроганов, рагу, гуляш и др.), после взвешивания извлекают с помощью пинцета кусочки мяса, смывая с них остатки соуса отмеренным количеством (25 - 30 см3) горячей воды и фильтровальной бумагой удаляют задержавшуюся на поверхности воду. Кусочки взвешивают и сравнивают массу их с выходом по рецептуре.
Воду, использованную для ополаскивания, добавляют к соусу и гарниру и измельчают их до однородной консистенции в размельчителе тканей. Общую массу гарнира и соуса определяют по разности между первоначальной массой блюда и массой основного продукта. Если масса мяса окажется заниженной, определяют содержание сухих веществ во всем блюде. В этом случае мясо нарезают, добавляют к соусу с гарниром, гомогенизируют все вместе в течение 5 мин.
В блюдах из запеченного рубленого мяса (рулеты) после взвешивания определяют массу основного изделия. Для этого с куска рулета или другого изделия счищают соус и вновь взвешивают его.
В изделиях, фаршированных мясным фаршем (запеканки, кабачки, перец фаршированные, голубцы), помимо массы основного изделия, определяют массу фарша, для чего взвешивают три изделия, отделяют от них фарш, взвешивают и сравнивают с выходом по рецептуре.
Каши рассыпчатые, блюда и гарниры из отварных, жареных, тушеных и запеченных овощей, бобовых и макаронных изделий, крупяные и овощные котлеты и биточки с жиром или соусом, блюда из муки и творога после подогрева и взвешивания гомогенизируют в размельчителе тканей с добавлением к ним горячей воды. Количество необходимой воды и продолжительность гомогенизации указаны в табл. 32. Дальнейший расчет содержания жира и сухих веществ ведут на массу блюда с водой*(18).
_________________
*(18) При отсутствии размельчителя растирают пробу в ступке.
Пельмени и вареники после взвешивания измельчают в размельчителе тканей, добавляя горячую воду. Содержание фарша определяют в полуфабрикате, отобранном одновременно на производстве.
Блины и оладьи для определения сухих веществ подготавливают, измельчая на мясорубке с последующей гомогенизацией. Для определения жира в мякише (за счет введения молока и яиц) с двух изделий снимают корочку толщиной 3 мм, а оставшийся мякиш измельчают на мясорубке и гомогенизируют.
Блинчики с фаршем очищают от сметаны или жира, взвешивают и определяют массу изделия. Для определения массы фарша взвешивают три изделия, отделяют фарш, взвешивают его и сравнивают с выходом по рецептуре. Отклонения от выхода допускаются в размере ±10 % (кроме блинчиков с творогом, где выход фарша регламентируется нормативно-технической документацией). Затем блинчики и фарш гомогенизируют отдельно и анализируют.
Соусы с наполнителями и без них измельчают в размельчителе тканей, в последнем случае - для равномерного распределения жира.
Количество добавляемой воды, продолжительность гомогенизации проб вторых блюд и гарниров и плотность серной кислоты, используемой для сжигания навески при определении содержания жира методом Гербера
|
Наименование изделий |
Количество воды, см3 на 100 г блюда |
Продолжительность гомогенизации, мин |
Плотность серной кислоты |
|
Рис откидной, каши рассыпчатые |
100 |
3 - 4 |
1,65 |
|
Макароны и лапша |
100 |
2 - 3 |
1,65 |
|
Крупяные котлеты, биточки, запеканки |
100 |
2 - 3 |
1,65 |
|
Сырники, запеканки, пудинги творожные |
50 |
3 - 4 |
1,82 |
|
Пюре картофельное |
10 - 15 |
1 - 2 |
1,65 |
|
Капуста тушеная |
50 - 70 |
5 - 6 |
1,7 |
|
Овощные котлеты, биточки, запеканки |
100 |
2 - 3 |
1,65 |
|
Голубцы, кабачки, фаршированные мясом |
50 - 70 |
2 - 3 |
1,65 |
|
Пельмени, оладьи, вареники, блины, блинчики |
100 |
2 - 3 |
1,65 |
Примечание. Учитывается количество воды, расходуемое на ополаскивание посуды, в которой доставлено блюдо, обмывание основного изделия от соуса и добавляемое на измельчение пробы.
4.4.1. Массовые доли сухих веществ и жира
Массовую долю сухих веществ определяют высушиванием подготовленной навески в сушильном шкафу или аппарате ВЧ (см. с. 8, 15), жира - методами Гербера или весовым с экстракцией жира в микроизмельчителе. Результаты анализов по этим показателям сравнивают с расчетными данными (см. п. 7.3).
4.4.2. Определение молока и сахара в крупяных изделиях
Аппаратура, материалы, реактивы. Аппаратура и материалы те же, что указаны на с. 159; реактивы: сульфат цинка, раствор массовой концентрации 150 г/дм3; гидроксид натрия, растворы массовой концентрации 40 г/дм3 и 100 г/дм3; метиловый красный, водно-спиртовой раствор массовой концентрации 2 г/дм3; соляная кислота, раствор с массовой долей 20 %.
Проведение испытания. Навеску гомогенизированной пробы массой 25 - 30 г взвешивают с точностью до 0,01 г в стеклянном стакане вместимостью 100 см3, добавляют 25 - 30 см3 воды, растирают стеклянной палочкой и переносят количественно в мерную колбу вместимостью 250 см3. Стакан ополаскивают несколько раз дистиллированной водой, сливая ее в мерную колбу. Вода должна занимать не более 2/3 объема мерной колбы. Мерную колбу встряхивают в течение 5 мин, затем приливают 10 см3 раствора сульфата цинка и 10 см3 раствора гидроксида натрия массовой концентрации 40 г/дм3, перемешивают, доводят дистиллированной водой до метки и оставляют стоять на 15 мин., после чего фильтруют в сухую колбу. В полученном фильтрате определяют редуцирующие сахара перманганатным методом по Бертрану*(19).
Для расчета содержания лактозы и определения количества молока количество меди, восстановленное редуцирующими сахарами, находящимися в растворе до инверсии сахарозы, пересчитывают на лактозу, пользуясь табл. 14.
Массу лактозы в порции крупяного изделия (г) вычисляют по формуле (19).
Количество молока в порции изделия (У, г) рассчитывают по формуле (75), где Х - масса лактозы в порции крупяного изделия, г.
Остальные обозначения, как в формуле (75).
Для определения массовой доли сахарозы определяют сумму редуцирующих сахаров до и после инверсии. Инверсию сахарозы проводят, как указано на с. 147.
Массовую долю сахарозы (S, г на порцию) в крупяных изделиях рассчитывают по формуле (21).
_______________
*(19) Параллельно с изделием из крупы определяют массовую долю редуцирующих сахаров в молоке, используемом для приготовления изделия, и пересчитывают их на лактозу.
4.4.3. Определение сахара в творожных изделиях
Аппаратура, материалы, реактивы. Аппаратура и материалы те же, что указаны на с. 47, 146; реактивы: сульфат меди, раствор 69,28 г в 1 дм3 дистиллированной воды; гидроксид натрия, раствор концентрации 1 моль/дм3; соляная кислота плотностью 1,19, раствор концентрации 7,3 моль/дм3; метиловый оранжевый, раствор массовой концентрации 1 г/дм3.
Проведение испытания. Навеску гомогенизированной пробы творожного изделия массой 25 г растирают в ступке или химическом стакане с небольшим количеством воды. Полученную суспензию количественно переносят в мерную колбу вместимостью 250 см3, смывая частицы в колбу дистиллированной водой, так, чтобы объем воды в колбе не превышал 2/3 ее объема. Для осаждения несахаров в колбу добавляют 5 см3 раствора сульфата меди и 2 см3 раствора гидроксида натрия. Содержимое колбы перемешивают и настаивают 5 мин. Если жидкость над осадком окажется мутной, в колбу следует добавить несколько капель раствора сульфата меди. Когда над осадком образуется прозрачный слой жидкости, колбу доливают водой до метки, перемешивают и оставляют на 20 - 30 мин, после чего фильтруют в сухую колбу (первые 25 см3 фильтрата отбрасывают).
Массовую долю редуцирующих сахаров в фильтрате определяют цианидным методом.
Для определения сахарозы проводят ее гидролиз. Для этого 25 см3 фильтрата пипеткой переносят в коническую колбу вместимостью 250 см3, закрывают ее пробкой с пропущенным через нее термометром так, чтобы ртутный резервуар находился в жидкости, и нагревают на водяной бане до температуры 65 ± 3 °С. Приоткрыв пробку, в колбу приливают 2,5 см3 раствора соляной кислоты, перемешивают и выдерживают на бане 10 мин при температуре 68 ± 2 °C. Затем колбу быстро охлаждают до 20 ± 2 °C, содержимое переносят количественно в мерную колбу на 100 см3, добавляют одну каплю метилоранжа и нейтрализуют гидроксидом натрия до слабокислой реакции. Содержимое мерной колбы доводят водой до метки. В полученном растворе цианидным методом определяют массовую долю редуцирующих сахаров после инверсии сахарозы.
Массовую долю сахарозы рассчитывают по формуле (21).
4.4.4. Определение содержания муки или манной крупы в творожных изделиях
Количество муки или манной крупы в творожных изделиях определяют по содержанию в них крахмала (см. с. 77).
4.4.5. Определение содержания хлеба в рубленых мясных и рыбных изделиях
Количество хлеба в рубленых мясных и рыбных изделиях определяют по содержанию в них крахмала (см. с. 74).
4.4.6. Определение количества мяса в кулинарных изделиях из рубленого мяса
Проведение испытания. Порцию изделия дважды пропускают через мясорубку и пробу массой 10 г отбирают в фарфоровую чашку. Далее экстрагируют пробу 200 см3 дистиллированной воды и анализируют, как указано на с. 138.
Обработка результатов. Содержание мяса в порции (Х, г) исследуемого образца определяют по формуле
|
|
(75) |
Обозначения те же, что и в формуле (70а).
Пример расчета. На испытание доставлены котлеты "Московские", приготовленные по II колонке рецептуры 660 Сборника рецептур, 1981 г.
По той же рецептуре из того же вида мяса (говядина) приготовлена порция контрольного полуфабриката с закладкой:
Говядина - 50 г (т.е. в формуле расчета С = 50)
Жир-сырец говяжий - 8,94 г
Лук репчатый - 1 г
Сухари - 4 г
Хлеб пшеничный - 14 г
Вода - 20,8 см3
Соль - 1,2 г
Перец - 0,06 г
-----------------
Масса полуфабриката 100 г. Полуфабрикат жарили на сковороде до готовности, расход жира для жарки - 5 г. Масса готовой жареной котлеты 81 г. Доставленный на испытание и приготовленный контрольный образец исследовали параллельно.
Результаты измерения оптической плотности: Дх = 0,455; Дк = 0,610.
Содержание мяса в исследуемой котлете равно:
![]()
Заключение. При норме закладки 50,00 г допустимые 10-процентные отклонения составляют ±5,00 г, т.е. минимальное содержание мяса в порции равно 50 - 5 = 45 г.
Таким образом, обнаружено недовложение мяса, которое составляет 45 - 37,3 = 7,7 г на порцию котлет "Московских".
4.5. Сладкие блюда
4.5.1. Желированные и выпеченные сладкие блюда
Отбор проб и физико-химические показатели, по которым контролируют соблюдение норм вложения сырья в сладкие блюда, приведены в прил. 2.
Подготовка проб к испытаниям. Кисель после удаления поверхностной пленочки тщательно перемешивают.
Желе и муссы (без крупы) расплавляют на водяной бане, охлаждают и перемешивают.
Кремы хорошо перемешивают.
Кремы на желатине готовят к испытаниям, как желе и муссы.
Пробы сиропа, используемого для поливки мусса, только перемешивают.
Блинчики с фаршем очищают от сметаны или жира. Взвешивают, осторожно развертывают, собирают фарш в предварительно взвешенную посуду и взвешивают. Масса фарша должна быть не менее 90 % выхода. Выпеченные блинчики измельчают ножом, затем растирают в ступке до получения однородной массы.
Из пудингов и сладких каш удаляют включения (орехи, изюм, цукаты), оставшуюся часть взвешивают и тщательно растирают в ступке.
4.5.1.1. Массовые доли сухих веществ и жира
Массовую долю сухих веществ определяют высушиванием навески в сушильном шкафу (см. с. 8) или рефрактометрическим методом (см. с. 17), жира - методами Гербера или экстракционно-весовым (см. с. 31, 39). Результаты анализов по этим показателям сравнивают с расчетными данными (см. п. 7.3).
4.5.1.2. Массовая доля сахара
В сладких кашах, пудингах из сухарей и круп сахар определяют, как в кашах (см. с. 164), в творожном пудинге и фарше творожном - как в изделиях из творога (см. с. 165). В желированных сладких блюдах сахар можно определить химическим методом (по Бертрану или цианидным) или рефрактометрическим.
Рефрактометрический метод определения сахара в желированных сладких блюдах
Аппаратура, материалы, реактивы. Рефрактометр универсальный типа УРЛ, РЛУ, ИРФ-457; весы лабораторные; стаканы химические вместимостью 50 см3; колба мерная вместимостью 100 см3; колбы конические вместимостью 100 - 150 см3; палочки стеклянные; штатив для пробирок; пробирки стеклянные вместимостью 20 см3; бумага фильтровальная; термометр лабораторный с диапазоном измерения 0 - 100 °C с ценой деления 1 °C; марля; воронки; цилиндр мерный вместимостью 50 см3; кислота уксусная ледяная, раствор с массовой долей 12 %; индикатор универсальный; сульфат меди (II), раствор с массовой долей 7 %; гидроксид натрия, раствор концентрации 1 моль/дм3 гексацианоферрат (II) калия (желтая кровяная соль), раствор с массовой долей 15 %; сульфат цинка, раствор с массовой долей 30 %; вода дистиллированная.
Проведение испытания. Перед началом работы рефрактометр подготавливают в соответствии с прилагаемой к прибору инструкцией.
Навеску фруктового желе, самбука (табл. 33) взвешивают в стакане с точностью до 0,01 г, переносят небольшим количеством воды (60 см3) с температурой 50 - 55 °C в мерную колбу вместимостью 100 см3, затем раствор охлаждают, колбу доливают водой до метки, перемешивают, дают жидкости отстояться 10 - 15 мин и фильтруют через бумажный фильтр в сухую колбу.
При исследовании желе молочного, муссов, кремов, киселей навески изделий переносят 40 см3 теплой (50 °C) воды (для фруктово-ягодных киселей 25 - 30 см3, т.к. избыток воды мешает осаждению крахмала) в мерную колбу вместимостью 100 см3. Затем для осаждения несахаров и осветления раствора добавляют 10 см3 раствора сульфата меди (II) и 4 см3 раствора гидроксида натрия (эквивалентное соотношение лучше установить титрованием).
|
Сладкие блюда и горячие напитки |
Навеска, г |
|
Мусс плодово-ягодный, желе молочное, фруктовое |
30 |
|
Самбук |
15 |
|
Кисели плодово-ягодные, плодово-ягодные из концентрата, кисель молочный, кремы |
25 |
Если жидкость над осадком будет мутной, количество осадителей увеличивают, сохраняя пропорцию растворов сульфата меди (II) и гидроксида натрия 2,5:1. После появления над осадком прозрачного слоя жидкости содержимое колбы охлаждают, доводят до метки дистиллированной водой, перемешивают, дают жидкости отстояться 10 - 15 мин и фильтруют через бумажный фильтр в сухую колбу, а затем рефрактометрируют.
Обработка результатов. Массовую долю сахарозы (Х, %) рассчитывают по формуле
|
X = K∙(a - в)∙10000, |
(77) |
где:
a - показатель преломления испытуемого раствора;
в - показатель преломления дистиллированной воды (при 20 °C равен 1,3330);
K - коэффициент пересчета показателя преломления на массовую долю сахара в исследуемом растворе;
10000 - множитель, введенный для того, чтобы разность (a - в) была целым числом.
Коэффициент K определяют экспериментально по результатам исследования контрольного образца, приготовленного из сырья, отобранного одновременно с исследуемым образцом. Контрольный напиток готовят в количестве трех порций.
Коэффициент K рассчитывают по формуле
|
|
(78) |
где С - массовая доля сахара в напитке, %.
Определение сахара в желированных сладких блюдах перманганатным методом по Бертрану или цианидным методом
Аппаратура, материалы, реактивы. Те же, что указаны на с. 47, 55, 159.
Проведение испытания. Навеску подготовленной для исследования пробы (табл. 33) берут в стакан вместимостью 50 - 100 см3 с точностью до 0,01 г и количественно переносят 100 - 150 см3 теплой воды (для желе, муссов на желатине, кремов, самбуков температурой 70 °C, для киселей и муссов на манной крупе - 50 °C) в мерную колбу вместимостью 250 см3. Содержимое колбы перемешивают в течение 5 мин.
Для осаждения несахаров в колбу при исследовании плодово-ягодных киселей, желе, муссов добавляют по 2 см3 раствора гексацианоферрата (II) калия с массовой долей 15 % и раствора сульфата цинка с массовой долей 30 %; при анализе молочных киселей, желе, кремов добавляют по 3 см3 указанных осадителей. Содержимое колбы доводят до метки дистиллированной водой, хорошо перемешивают и оставляют на 20 мин для осаждения осадка. Надосадочная жидкость должна быть прозрачной. Ее фильтруют в сухую колбу. В фильтрате определяют редуцирующие сахара до инверсии перманганатным (по Бертрану) или цианидным методом.
При определении сахарозы проводят ее гидролиз, как указано на с. 147. В полученном растворе определяют редуцирующие сахара после инверсии сахарозы.
Определив массу редуцирующих сахаров до и после инверсии сахарозы, количество сахарозы (в г на порцию) рассчитывают по формуле (21).
4.5.1.3. Определение содержания молока в сладких блюдах
Полноту вложения молока в кисели, желе, пудинги и сладкие каши определяют по лактозе, как в молочных кашах (см. с. 164).
4.5.1.4. Определение содержания манной крупы в сладких блюдах Содержание манной крупы в муссах и творожном пудинге определяют по крахмалу (см. с. 77).
4.5.2. Компоты
Для определения плотной части компотов пять порций соединяют вместе, взвешивают, после чего отделяют плотную часть, пользуясь металлическим ситом с жестяным корпусом высотой 10 - 15 см или дуршлагом. Через 10 мин плотную часть взвешивают с точностью до 1 г.
Отклонение массы фруктов от выхода, предусмотренного рецептурой, допускается в размере ±10 %. Массу плотной части сравнивают с расчетными данными, полученными с учетом потерь при тепловой обработке, а для сухофруктов - увеличения массы при варке (Сборник рецептур блюд и кулинарных изделий для предприятий общественного питания, 1981 г.).
4.6. Напитки
Отбор проб и физико-химические показатели, по которым контролируют соблюдение норм вложения сырья, приведены в прил. 2.
4.6.1. Чай
4.6.1.1. Определение свежести настоя чая
Кипячение настоя чая приводит к потере аромата, прозрачности и ухудшению цвета: из оранжево-желтого он становится грязно-коричневым. Чай, подвергшийся кипячению, снимается с реализации и дальнейшему анализу не подлежит.
Аппаратура, материалы, реактивы. Песочная баня; пипетки вместимостью 1 см3 и 2 см3, градуированные; гексацианоферрат (III) калия, раствор с массовой долей 1 %; гидроксид натрия, раствор с массовой долей 40 %.
Проведение испытания. Готовят контрольный настой (заварку) по рец. 1008. Исследуемую и контрольную заварку охлаждают до комнатной температуры и фильтруют через бумажный фильтр.
Для определения свежести настоя в две пробирки наливают по 1 см3 профильтрованного испытуемого и контрольного настоя. К пробам добавляют по 2 см3 раствора гексацианоферрата (III) калия и раствора гидроксида натрия. Содержимое пробирок встряхивают и оставляют на 5 - 10 мин. При кипячении настоя или недовложении в него сухого чая жидкость в пробирке окрашивается в светло-желтый цвет, при вторичной заварке спитого чая - в лимонный; жидкость в контрольной пробирке - золотистая.
4.6.1.2. Определение экстрактивных веществ в настое чая (заварке) или напитке
Настой чая отфильтровывают и отбирают по 10 см3 в предварительно взвешенные металлические бюксы. Упарив досуха на песочной бане, остаток досушивают 0,5 ч в сушильном шкафу при температуре 105 °C и взвешивают.
Массовую долю экстрактивных веществ (Х, %) в настое (заварке) или напитке рассчитывают по формуле
|
|
(79) |
где:
K - коэффициент пересчета, равный для заварки 5, для напитка - 20;
m - масса сухого остатка в бюксе, г;
m1 - масса сухого чая на порцию, г.
Массовые доли экстрактивных веществ в чае различных сортов приведены в табл. 34.
Таблица 34
|
Наименование чая |
Массовая доля экстрактивных веществ в % к массе сухого чая |
|
|
сорта |
||
|
высший |
первый |
|
|
Грузинский |
32,0 |
29,2 |
|
Краснодарский |
33,9 |
29,4 |
|
Индийский |
36,5 |
30,8 |
|
Цейлонский |
33,2 |
30,1 |
|
Азербайджанский |
30,1 |
28,3 |
Если сорт чая, из которого приготовлена заварка или напиток, неизвестен, найденное содержание экстрактивных веществ должно быть не менее 28,3 %.
4.6.1.3. Определение крепости настоя чая по эталонам
Метод основан на визуальном сравнении цвета исследуемого чая-заварки (напитка) с цветом эталонных настоев с известным содержанием сухого чая (колориметрирование в ряду стандартов).
Метод пригоден для анализа напитков с сахаром или без сахара, но без примеси жженого сахара или соды.
Проведение испытания. Контрольный чай-заварку готовят из того же сорта сухого чая, из которого был приготовлен исследуемый чай-заварка. Для этого берут 10 г сухого чая с точностью до 0,01 г и 500 см3 воды. Заварку готовят в соответствии с технологическими требованиями, настаивают в течение 10 мин и охлаждают до температуры 20 °C.
Заварку фильтруют и, разбавляя ее дистиллированной или кипяченой водой, готовят контрольные растворы № 2 - 7 (табл. 35).
Таблица 35
Контрольные растворы для колориметрического определения количества сухого чая
|
№ контрольного раствора |
Разведение контрольной заварки в частях по объему |
Содержание сухого чая |
||
|
заварка |
вода |
в 50 см3 заварки, г |
в 200 см3 напитка, г |
|
|
1* |
1 |
0 |
2 |
- |
|
2 |
1 |
0 |
1 |
- |
|
3 |
3 |
1 |
0,75 |
- |
|
4 |
2 |
2 |
0,50 |
2,0 |
|
5 |
1 |
3 |
0,25 |
1,0 |
|
6 |
2 |
13 |
0,187 |
0,75 |
|
7 |
1 |
7 |
0,125 |
0,5 |
________________
* Контрольный раствор № 1 готовят из 20 г сухого чая и 500 см3 воды, контрольные растворы № 2 - 7 готовят из 10 г сухого чая и 500 см3 воды.
Контрольные растворы помещают в пробирки из бесцветного стекла, пробирки устанавливают в штативе. Исследуемый напиток (чай-заварка) сравнивают по цвету с контрольными растворами на фоне белой бумаги и по таблице находят содержание сухого чая.
Если цвет испытуемого напитка (чая-заварки) не совпадает точно ни с одним из контрольных напитков, то содержание сухого чая можно определить как среднее между двумя соседними контрольными растворами, которые по интенсивности окраски близки к исследуемому напитку (чаю-заварке).
Пример. Исследуемый чай-заварка, приготовленный по рец. 1008, соответствует по цвету контрольному раствору № 5. Следовательно, в 50 см3 чая-заварки содержится 0,25 г сухого чая.
Заключение. Исследуемый чай-заварка не соответствует рец. 1008, т.к. норма вложения сухого чая на 50 см3 заварки должна составлять 2 г (кол. I) или 1 г (кол. II, III).
4.6.1.4. Определение массы сухого чая, использованного при приготовлении напитка чай
Метод основан на цветной реакции (образование зеленой окраски) полифенольных соединений чая с ионами железа-3 в азотнокислой среде. Величина оптической плотности при длине волны 630 - 670 нм линейно зависит от массы сухого чая.
Сахар, жженый сахар, питьевая сода, если они были добавлены при приготовлении заварки, не мешают анализу.
Аппаратура, материалы, реактивы. Весы лабораторные; фотоэлектроколориметр, обеспечивающий измерения в интервалах волн 315 - 670 нм; плитка электрическая; чайник заварочный фарфоровый вместимостью 500 см3; пипетки вместимостью 5 см3, 10 см3; цилиндры вместимостью 10, 25 и 250 см3; колбы мерные вместимостью 500 см3; колбы конические вместимостью 50, 250 см3; воронки стеклянные; бумага фильтровальная; азотная кислота, раствор с массовой долей 1,5 %: к 200 см3 дистиллированной воды, помещенным в мерную колбу вместимостью 500 см3, вливают 8 см3 концентрированной азотной кислоты и доводят объем водой до метки, перемешивают; реактив: раствор нитрата железа (III) массовой долей 1 % в растворе азотной кислоты с массовой долей 1,5 % (5 г нитрата железа (III) растворить в 500 см3 раствора азотной кислоты с массовой долей 1,5 % и перемешать).
Проведение испытания. Для проведения испытания из того же сорта сухого чая готовят в фарфоровом чайнике 200 см3 контрольной заварки. Для этого берут 8 г сухого чая и 220 см3 воды (рец. 1008, кол. I) или 4 г сухого чая и 216 см3 воды (рец. 1008, кол. II, III). Воду отмеривают цилиндром и доводят в колбе до кипения, а затем используют для заварки. Готовую заварку отфильтровывают и доводят до температуры 20 °C. В коническую колбу вместимостью 250 см3 вносят цилиндром 50 см3 приготовленного чая-заварки и 150 см3; воды при 20 °C, перемешивают*(20). Получается порция 200 см3 контрольного напитка, в которой эквивалентное содержание сухого чая равно С = 2,0 г (кол. I) или 1,0 г (кол. II, III).
_______________
*(20) Если исследуемый напиток содержит сахар, то такую же массу сахара добавляют и в контрольный напиток, затем перемешивают до растворения сахара.
Затем исследуемый (доставленный на испытание) и приготовленный контрольный напиток анализируют параллельно по следующей методике. Порцию напитка (200 см3) доводят до температуры 20 °C и фильтруют в сухую коническую колбу.
В три сухие конические колбы вместимостью 50 см3 вносят пипетками по 10 см3 профильтрованного напитка и 10 см3 дистиллированной воды и перемешивают. Затем в первую колбу приливают пипеткой 5 см3 реактива нитрата железа (III), быстро перемешивают и немедленно измеряют оптическую плотность полученного зеленого раствора на заранее включенном фотоэлектроколориметре при длине волны 630 - 670 нм в кювете толщиной 20 мм. В кюветы сравнения наливают дистиллированную воду. Источник света в приборах ФЭК-56, ФЭК-56М - обычная лампа накаливания. Указанную операцию проводят по очереди с остальными двумя колбами. За окончательный результат измерения оптической плотности принимают среднее арифметическое.
Обработка результатов испытания. Эквивалентную массу сухого чая (Х, г) в пересчете на порцию 200 см3 исследуемого напитка вычисляют по формуле
|
|
(80) |
где:
Дх и Дк - соответственно величины оптической плотности исследуемого и контрольного напитков;
m - масса сухого чая в пересчете на порцию контрольного напитка, г (m = 2,0 г по кол. I и m = 1,0 г по кол. II, III рец. 1008).
Пример расчета. На анализ доставлен напиток чай, приготовленный по рец. 1008, кол. I. По той же рец. из 8,0 г того же сорта чая и 220 см3 воды приготовлен чай-заварка, а из 50 см3 этого чая-заварки и 150 см3 воды - порция 200 см3 контрольного напитка (эквивалентная масса сухого чая в расчете на эту порцию 2,0 г).
Оба напитка, исследованный и контрольный, анализировались параллельно, как описано выше. Результаты измерения оптической плотности Дх = 0,480; Дк = 0,840.
Масса сухого чая в пересчете на порцию исследуемого напитка равна:
![]()
Заключение. Исследуемый напиток не соответствует рецептуре. При норме вложения 2,0 г сухого чая допустимые 10-процентные отклонения (производственные потери, погрешности взвешивания и анализа) составляют ±0,20 г. Минимальная масса сухого чая 1,80 г (2,00 - 0,20). Недовложение сухого чая при приготовлении напитка составляет 0,66 г (1,80 - 1,14) в пересчете на порцию 200 см3.
4.6.1.5. Обнаружение питьевой соды в чае-заварке
Заварки, приготовленные в соответствии с рецептурой, имеют слабокислую среду, величину pH от 5,20 до 6,70. Добавление соды создает щелочную среду (pH от 7,20 до 8,00), в которой усиливается окисление катехинов чая и, как следствие, возрастает интенсивность окраски заварки. При этом может маскироваться недовложение сухого чая или использование разваренного чайного листа (спитого чая).
Проведение испытания. Чай-заварку доводят до комнатной температуры и измеряют величину pH на pH-метре. Для ускоренного определения каплю охлажденной до комнатной температуры заварки наносят на полоску универсальной индикаторной бумаги. Зеленая окраска бумаги свидетельствует о наличии соды.
4.6.1.6. Обнаружение жженого сахара в чае-заварке
Метод обнаружения жженого сахара основан на том, что дубильные вещества, содержащиеся в чае, дают осадки с солями некоторых металлов, а растворы жженого сахара таких осадков не образуют.
Реактивы: ацетат меди (II), раствор с массовой долей 9 %.
Проведение испытания. В сухую пробирку наливают 5 см3 настоя чая с температурой 18 ± 2 °C, добавляют 2 см3 раствора ацетата меди (II), тщательно перемешав содержимое пробирки, оставляют на 15 - 20 мин. По цвету жидкости, наличию или отсутствию осадка делают заключение о присутствии в настое жженого сахара (табл. 36).
Таблица 36
Характеристика настоя чая
|
Образцы настоя |
Наличие осадка |
Цвет жидкости над осадком |
|
Настой чая без добавления жженого сахара |
Есть |
Зеленоватый |
|
Настой чая с добавлением жженого сахара |
Есть |
Зеленовато-бурый |
|
Раствор жженого сахара |
Нет |
Золотисто-коричневый |
4.6.1.7. Массовая доля сухих веществ в чае
Определение сухих веществ в чае проводят рефрактометрическим методом, как указано на с. 17.
4.6.2. Кофе, какао
4.6.2.1. Обнаружение замены натурального кофе кофейным напитком
В состав кофейных напитков входят зерновые продукты: ячмень, овес, рожь. Содержащийся в них крахмал можно обнаружить специфической реакцией его с йодом. На этом основан метод обнаружения замены натурального кофе кофейным напитком.
Аппаратура, материалы, реактивы. Выпарительная чашка диаметром 5 - 7 см; капельница; палочка стеклянная; раствор Люголя.
Проведение испытания. В выпарительную чашку наливают 1 см3 профильтрованного напитка, разбавляют 5 см3 дистиллированной воды, перемешивают стеклянной палочкой, добавляют две-три капли раствора Люголя. Если кофе был приготовлен с добавлением кофейного напитка, жидкость окрасится в фиолетово-синий цвет, переходящий через 5 - 10 с в напитках с молочными продуктами в светло-коричневый. При отсутствии кофейного напитка появившаяся желтоватая окраска постепенно исчезает.
4.6.2.2. Определение массы натурального кофе в напитках*(21)
__________________
*(21) Метод разработан в Харьковском институте общественного питания В.П. Максимцом, Э.Ф. Кравченко, Л.И. Осинской.
Метод основан на линейной зависимости величины оптической плотности при длине волны 310 - 320 нм от массы натурального кофе в напитках. В указанном интервале длин волн УФ-лучи поглощает хлорогеновая кислота, которая содержится в натуральном кофе и отсутствует в его заменителях.
Метод предназначен для определения полноты вложения кофе в напитки "Кофе черный", "Кофе черный с молоком или сливками", "Кофе на молоке", "Кофе на молоке сгущенном", кофе из консервов "Кофе натуральный со сгущенным молоком и сахаром", "Кофе черный с мороженым (гляссе)".
Аппаратура, материалы, реактивы. Весы лабораторные; фотоэлектроколориметр, обеспечивающий измерения в интервале волн 315 - 630 нм с погрешностью не более 0,1Д (по оптической плотности); плитка электрическая; колбы мерные вместимостью 50, 200 см3; пипетки вместимостью 1, 2 см3; колбы конические вместимостью 250 см3; цилиндры вместимостью 10, 25, 100 и 250 см3; пробирки химические; стеклянные воронки; делительные воронки вместимостью 100 - 250 см3; бумага фильтровальная, медленно фильтрующая "синяя лента"; гексан или петролейный эфир; кислота трихлоруксусная с массовой долей 30 %.
Проведение испытания. Для проведения испытания готовят контрольный напиток из того же сорта кофе и по той же рецептуре, что и исследуемый напиток.
Оба напитка, исследуемый и контрольный, доводят до температуры 20 °C и измеряют их объем.
"Кофе черный" с сахаром и без сахара. Напиток фильтруют через сухой складчатый двойной фильтр из медленно фильтрующей бумаги "синяя лента" в сухую пробирку. Пипеткой отбирают 1 см3 прозрачного фильтрата и переносят в мерную колбу вместимостью 200 см3, доливают дистиллированной водой до метки и тщательно перемешивают. Величину оптической плотности приготовленных водных растворов (контрольного и исследуемого) измеряют при длине волны 315 нм в кювете на 10 мм. Источник света в приборах ФЭК-56, ФЭК-56М - ртутная лампа. В кюветы сравнения наливают дистиллированную воду.
Обработку результатов проводят, как указано на с. 179.
Кофе с молочными продуктами. Исследуемый и контрольный напитки доводят до температуры 20 °C и тщательно перемешивают. Напитки прокисшие и со свернувшимся молоком анализу не подлежат.
В делительную воронку вносят последовательно пипетками 20 см3 дистиллированной воды, 10 см3 напитка, перемешивают, приливают цилиндром 10 см3 гексана или петролейного эфира и пипеткой 10 см3 раствора трихлоруксусной кислоты с массовой долей 30 %. Воронку закрывают пробкой и энергично встряхивают в течение 5 - 10 с, при этом осаждаются белки и экстрагируется жир. Пробку удаляют и после отстаивания в течение 3 - 5 мин нижний (водный) слой фильтруют через сухой двойной фильтр из бумаги "синяя лента" в сухую пробирку. Пипеткой отбирают 2 см3 прозрачного фильтрата, переносят в мерную колбу вместимостью 50 см3, доводят до метки дистиллированной водой и тщательно перемешивают. Измерение величины оптической плотности описано выше.
Обработка результатов. Массу кофе (Х, г) в порции исследуемого напитка рассчитывают по формуле
где:
m - масса кофе в порции контрольного напитка, г;
Vx, Vк - соответственно объемы исследуемого и контрольного напитков, см3;
Дх и Дк - соответственно величины оптической плотности для исследуемого и контрольного напитков.
При расчете массы кофе в напитках с молочными продуктами вычисленную по формуле величину Х нужно умножить на поправочный коэффициент K. Масса Х1 кофе в порции напитка с молочными продуктами равна
|
Х1 = Х∙K. |
(82) |
Величины коэффициента K находят по табл. 37.
|
Вычисленная по формуле величина X |
Поправочный коэффициент K |
|
от 0,5 до 2,9 |
0,75 |
|
от 3,0 до 4,9 |
0,85 |
|
от 5,0 г и более |
0,95 |
Примечание. При анализе напитков кофе, приготовленных из концентратов "Кофе натуральный со сгущенным молоком и сахаром", поправочный коэффициент в расчете не принимается, т.к. вычисляется не масса кофе, а масса консервов в напитке.
Пример расчета 1. На анализ доставлен напиток "Кофе черный" без сахара (порция 200 см3), приготовленный по рец. 1014, 2-й вариант, кол. II, III.
По той же рецептуре из того же сорта молотого кофе приготовлена методом наплитной варки порция 200 см3 контрольного напитка с закладкой кофе 8,0 г.
Оба напитка анализировались параллельно. Результаты измерения оптической плотности: Дх = 0,650; Дк = 0,680. Масса кофе в порции исследуемого напитка равна:
![]()
(Величины объемов Vx и Vк равны, поэтому исключены из расчета.)
Заключение. Исследуемый напиток соответствует рецептуре. Разность 0,35 г (8,00 - 7,65) находится в пределах допустимых 10-процентных отклонений, рассчитанных на производственные потери при изготовлении и порционировании напитков, погрешности при взвешивании и анализе. В данном случае, при закладке кофе 8 г, отклонения составляют ±0,80 г.
Пример расчета 2. На анализ доставлен напиток "Кофе на молоке" (рец. 1017, кол. III). По той же рецептуре из того же сорта молотого кофе приготовлена порция 200 см3 контрольного напитка с закладкой 1/5 от рецептуры, т.е.: кофе натуральный - 6 г; молоко - 50 см3; вода - 168 см3; сахар - 20 г. Оба напитка анализировались параллельно. Результаты измерения оптической плотности при 315 нм: Дх = 0,610; Дк = 0,725. Рассчитывают массу кофе в порции (Х, г) исследуемого напитка
![]()
Затем найденную величину Х умножаем на поправочный коэффициент K, равный 0,95 (см. табл. 37).
Таким образом, масса кофе в порции исследуемого напитка равна:
Х1 = 5,05∙0,95 = 4,80 г.
Заключение. Исследуемый напиток не соответствует рецептуре. При закладке кофе по норме 6 г допустимые отклонения составляют ±0,60 г, а минимальное содержание кофе в порции 5,54 г (6,00 - 0,60). Недовложение кофе на порцию равно 0,74 (5,54 - 4,80).
4.6.2.3. Определение массы порошка какао в напитках какао с молочными продуктами*(22)
_______________
*(22) Разработан в Харьковском институте общественного питания В.П. Максимцом, Э.Ф. Кравченко, Л.И. Осинской, Ж.В. Белега.
Метод основан на линейной зависимости интенсивности розовой окраски водной вытяжки из напитка от массы порошка какао. Измерение оптической плотности проводят при длине волны 440 нм (синий светофильтр) после предварительного осаждения белков и экстракции жира.
Аппаратура, материалы, реактивы. Фотоэлектроколориметр, обеспечивающий измерения в интервале волн 315 - 630 мкм с погрешностью не более 0,1Д (по оптической плотности); весы лабораторные; плитка электрическая; центрифуга лабораторная; колбы конические вместимостью 250, 500 см3; пипетки вместимостью 5, 10 см3; цилиндры вместимостью 10, 100 см3, 250 см3; пробирки химические; стеклянные воронки; делительные воронки вместимостью 100 - 250 см3; бумага фильтровальная, медленно фильтрующая "синяя лента"; гексан или петролейный эфир; кислота трихлоруксусная, раствор с массовой долей 40 %.
Проведение испытания. Готовят порцию 200 см3 контрольного напитка какао из того же сорта порошка какао, по той же рецептуре, что и исследуемый напиток. Оба напитка, исследуемый и контрольный, доводят до комнатной температуры и анализируют параллельно, как описано ниже.
Напиток тщательно перемешивают и отливают 15 - 20 см3 в центрифужную пробирку. Центрифугируют при 1000 - 3000 об/мин в течение 5 - 7 мин. После центрифугирования стеклянной палочкой удаляют слой жира с поверхности и пипеткой отбирают 5 см3 центрифугата над коричневым осадком, не взмучивая его.
В сухую делительную воронку вносят последовательно пипетками 10 см3 дистиллированной воды, 5 см3 центрифугата, приливают цилиндром 10 см3 гексана или петролейного эфира и пипеткой 10 см3 раствора трихлоруксусной кислоты. Делительную воронку закрывают пробкой и смесь очень сильно встряхивают в течение 1 - 2 мин, затем удаляют пробку и дают смеси отстояться 2 - 3 мин. Встряхивание смеси в делительной воронке должно быть очень энергичным, чтобы обеспечить полноту осаждения белков и экстракции жира, иначе при последующем фильтровании получаются мутные фильтраты, непригодные для фотометрирования. Нижний (водный) слой розового цвета фильтруют через сухой двойной фильтр из бумаги "синяя лента" в сухую кювету фотоэлектроколориметра (кювета 20 мм). Фильтрат должен быть прозрачным.
Оптическую плотность измеряют против дистиллированной воды при длине волны 440 нм. Источник света в ФЭК-56, ФЭК-56М - лампа накаливания.
Обработка результатов испытания. Массу порошка какао (Х, г) в порции напитка находят по формуле
|
|
(83) |
где:
Дх и Дк - соответственно величины оптической плотности Д для исследуемого и контрольного напитка;
m - масса какао-порошка в порции контрольного напитка, г.
Если объемы исследуемого и контрольного напитков различны, то расчет ведут по формуле (81), где
m - масса порошка какао в порции контрольного напитка, г.
Пример расчета. На анализ доставлен напиток "Какао с молоком" (рец. 1025, кол. II). По той же рецептуре, из того же сорта порошка какао приготовлена порция 200 см3 контрольного напитка с закладкой на порцию (1/5 рецептуры): какао-порошка - 5 г; молока - 130 см3; воды - 80 см3; сахара - 25 г.
Результаты измерения величины оптической плотности при 440 нм: исследуемый напиток - Дх = 0,265; контрольный напиток - Дк = 0,410.
Масса (Х, г) порошка какао в порции исследуемого напитка равна:
![]()
Заключение. При норме закладки 5,00 г допустимые отклонения при приготовлении, порционировании и погрешностях взвешивания и анализа составляют ±0,50 г, т.е. минимальное содержание порошка какао в порции равно 4,50 г (5,00 - 0,50). Исследуемый напиток не соответствует рецептуре: недовложение порошка какао на порцию 200 см3 составляет 1,27 г (4,50 - 3,23).
4.6.2.4. Определение количества натурального кофе в напитке "Кофе черный" без сахара по массовой доле экстрактивных веществ
Массовую долю экстрактивных веществ в напитке определяют рефрактометрически.
Аппаратура, материалы, реактивы. См. с. 17.
Проведение испытания. Готовят контрольный напиток из зерен кофе, отобранных на месте приготовления напитка, по той же рецептуре, что и исследуемый напиток. Если исследуемый напиток приготовлен из молотого кофе, упакованного в пачки или пакеты, контрольный напиток готовят из кофе в пачках или пакетах заводской упаковки.
При испытаниях напитка, приготовленного в кофеварках типа "Экспресс", контрольный напиток готовят в той же кофеварке и в той же ячейке, что и исследуемый, для соблюдения одинаковых условий экстракции кофе.
Исследуемый и контрольный напитки охлаждают до комнатной температуры, измеряют их объем и фильтруют в сухие колбы. В фильтратах определяют показатель преломления и рассчитывают массовую долю сухих (экстрактивных) веществ в % (см. выше). Результаты испытаний исследуемого и контрольного образцов сравнивают. Допускаемые отклонения ±0,2 %.
Если контрольный образец приготовить не представляется возможным, результат испытания сравнивают с расчетным по рецептуре, принимая за минимально допустимую норму среднее значение содержания экстрактивных веществ в кофе по ГОСТ 6805-83 "Кофе натуральный жареный", равное 25 % с учетом влажности кофе (не более 7%)*(23).
_________________
*(23) При варке кофе в наплитной посуде, электрокофеварках и полуавтоматических кофеварках типа "Экспресс" содержание экстрактивных веществ равно в среднем также 25 %.
4.6.2.5. Определение массовой доли сухих веществ в кофе (какао) с молоком
Массовую долю сухих веществ определяют рефрактометрически без предварительного осаждения белков.
Проведение испытаний. Температуру напитка доводят до 20 °C, измеряют объем поступившей порции и фильтруют напиток через вату или бумажный фильтр в сухую коническую колбу, фильтрат рефрактометрируют, как указано на с. 17.
Данные анализа сравнивают с массовой долей сухих веществ по рецептуре (Х, %), рассчитанной по формуле
|
|
|
|
|
|
|
или Х = 0,5∙а + 0,005(СД + ВЕ), |
(84) |
где:
Х - массовая доля сухих веществ, рассчитанная по рецептуре, %;
a - масса сахара в порции напитка по рецептуре, г;
С - массовая доля экстрактивных (растворимых в воде) сухих веществ по отношению к количеству кофе или какао, взятых по рецептуре, с учетом их влажности, % (для кофе принимается равной 25 %, для какао - 35 %);
Д - количество кофе или какао, указанное в рецептуре, г;
В - массовая доля сухих веществ в молоке, видимых по рефрактометру (принимается равной 10 %);
Е - количество молока, указанное в рецептуре для приготовления порции напитка, г.
Допустимые отклонения в содержании сухих веществ с учетом потерь при производстве и порционировании напитков составляют для кофе ±1,5 %, для какао - ±2,0 %.
При использовании сгущенного молока для приготовления напитков 50 г его равноценны 25 г сахара и 100 г молока жирностью 3,2 %.
Пример расчета 1. По рец. 1014 (1-й вариант) на
порцию 100 см3 закладка кофе равна 6 г. При влажности кофе не более
7 % масса сухого кофе в порции равна 5,58 г ![]() . Экстрактивных веществ в кофе (ГОСТ
6805-83) 25 %, или 1,4 г
. Экстрактивных веществ в кофе (ГОСТ
6805-83) 25 %, или 1,4 г ![]() .
.
С учетом допустимых потерь минимально допустимое содержание экстрактивных веществ составляет 1,26 г.
Пример расчета 2. По рец. 1017 (кол. III) на одну порцию кофе с молоком (выход 200 см3) вложение продуктов массой нетто, г: кофе натуральный - 6, сахар - 20, молоко - 50.
Масса порошка кофе в пересчете на сухое вещество при
влажности кофе, равной 7 %: 5,58 г ![]() .
.
Расчетное содержание сухих веществ: 0,5 × 20 + 0,005∙(25 × 6 + 10 × 50) = 13,25 %.
При допустимом отклонении ±1,5 % минимально допустимое содержание сухих веществ 11,75 %, или в одной порции 23,5 г.
4.6.2.6. Определение сахара в кофе черном, чае, кофе и какао с молоком
Массовую долю сахара в чае, кофе черном, кофе и какао с молоком определяют рефрактометрическим методом.
Аппаратура, материалы, реактивы. Рефрактометр универсальный (УРЛ, РДУ, ИРФ-457); пипетка вместимостью 10 см3; колба коническая вместимостью 100 - 150 см3; колба мерная вместимостью 100 см3; пробирки; уксусная кислота, раствор с массовой долей 12 %; гексацианоферрат (II) калия, раствор с массовой долей 15 %; сульфат цинка, раствор с массовой долей 30 %.
Проведение испытания. Температуру напитков перед испытанием доводят до 20 °C, измеряют объем поступивших порций и фильтруют их через вату или бумажный фильтр в сухую коническую колбу. Параллельно готовят контрольный напиток по той же рецептуре, что и исследуемый, строго соблюдая технологию приготовления.
Кофе и какао с молоком (цельным или сгущенным) в количестве 10 - 15 см3 переносят пипеткой в коническую колбу и осаждают белки, добавляя шесть-семь капель раствора уксусной кислоты с массовой долей 12 % до выпадения белка крупными хлопьями (pH = 5 проверяют универсальным индикатором). Надосадочную жидкость фильтруют через сухой складчатый фильтр в сухую пробирку.
Чай и кофе черный с сахаром в количестве 20 - 25 см3 переносят в мерную колбу на 100 см3, несахара осаждают, добавляя по 1,5 см3 раствора гексацианоферрата (II) калия с массовой долей 15 % и раствора сульфата цинка с массовой долей 30 %, доводят содержимое колбы до метки, перемешивают, дают жидкости отстояться и фильтруют через бумажный фильтр в сухую колбу.
Подготовленные растворы (исследуемый и контрольный) рефрактометрируют и рассчитывают массовую долю сахара (Х, %) по формуле
|
Х = (а - б)∙1000∙K, |
(85) |
где:
a - показатель преломления испытуемого раствора;
б - показатель преломления дистиллированной воды (при 20 °C равен 1,3330);
K - коэффициент пересчета показателя преломления на массовую долю сахара в исследуемом растворе;
10000 - множитель, введенный для того, чтобы разность (a - б) была целым числом.
Коэффициент (K) определяют экспериментально по результатам исследования контрольного образца, приготовленного из сырья, отобранного одновременно с исследуемым образцом. Контрольный напиток готовят в количестве трех порций.
Коэффициент (K) рассчитывают по формуле
|
|
(86) |
где С - массовая доля сахара в напитке, %.
Результаты рассчитывают с точностью до 0,1 % и сравнивают с минимально допустимым результатом. Допустимые отклонения в результатах анализа контрольного и исследуемого образцов ±0,2 %.
Количество сахара (Х2, г на порцию напитка) определяют по формуле
|
|
(87) |
где:
С - массовая доля сахара в напитке, %;
m - масса порции напитка, г.
4.6.2.7. Определение количества молока в кофе (какао) с молоком Вложение молока в напитки контролируют по содержанию лактозы. Одновременно определяют массовую долю лактозы в молоке, используемом для приготовления напитка. Если исследовать его не представляется возможным, содержание лактозы принимается равным 4,7 %.
Аппаратура, материалы, реактивы. Те же, что на с. 58.
Проведение испытания. Напиток (45 см3) переносят в мерную колбу вместимостью 250 см3, добавляют 3 - 4 см3 раствора сульфата цинка массовой концентрации 200 г/дм3 и 1,5 - 2 см3 раствора гидроксида натрия концентрации 2,5 моль/дм3 для осаждения белков. Раствор в колбе тщательно перемешивают, доводят дистиллированной водой до метки и через 10 мин фильтруют в сухую коническую колбу. В фильтрате определяют лактозу ускоренным цианидным методом (см. с. 57).
Количество молока (У, г) в порции напитка рассчитывают по формуле
|
|
(88) |
где:
А - масса лактозы в напитке, г;
Х1 - количество лактозы в молоке, используемом для приготовления напитка.
Для приготовления напитка молоко берут по объему, поэтому для определения количества молока (в см3) найденную величину надо разделить на максимально допустимую плотность молока, равную 1,032 г/см3.
При исследовании напитков, приготовленных со сгущенным молоком, количество сгущенного молока (Х, г) находят по формуле
|
|
(89) |
где:
m - масса лактозы, найденная в порции напитка, г;
12,5 - массовая доля лактозы в консервах "Молоко сгущенное с сахаром", %.
Пример расчета. Определяли содержимое молока в кофе, приготовленном по рецептуре: кофе натуральный с цикорием - 8 г, сахар - 25 г, молоко - 75 см3.
Объем напитка составил 200 см3. 45 см3 напитка перенесли в мерную колбу вместимостью 250 см3. На титрование 10 см3 раствора гексацианоферрата (II) калия затрачено 4,8 см3 фильтрата. Количество лактозы в напитке равно
![]()
Молока, использованного для приготовления напитка, на
предприятии не оказалось. Масса молока на порцию напитка 59,9 г ![]() или 57 см3
или 57 см3![]() .
.
Заключение. С учетом допустимых отклонений (±10 %) содержание молока в порции напитка занижено на 10,5 см3.
4.6.3. Шоколад
4.6.3.1. Определение количества шоколада в напитке "Шоколад"*(24)
Аппаратура, материалы, реактивы. Фотоэлектроколориметр, обеспечивающий измерения в интервалах волн 315 - 670 нм; бумага фильтровальная, медленно фильтрующая "синяя лента"; гексан или петролейный эфир; раствор трихлоруксусной кислоты с массовой долей 40 %.
_____________
*(24) Метод разработан Харьковским институтом общественного питания.
Проведение испытания. Порцию напитка (200 см3) доводят до температуры 20 °C и после тщательного перемешивания 25 см3 напитка цилиндром переносят в сухую делительную воронку. Наливают цилиндром 10 см3 гексана или петролейного эфира и добавляют пипеткой 10 см3 раствора трихлоруксусной кислоты. Смесь встряхивают в течение 5 - 7 с и оставляют для отстаивания на 15 - 20 мин для разделения ее на гексановую и водную фракции. При сокращении времени отстаивания не удается получить прозрачный фильтрат, и анализ следует производить заново. Нижний (водный) слой розового цвета фильтруют через однослойный фильтр из бумаги "синяя лента" в кювету (20 мм).
Измеряют оптическую плотность фильтрата при длине волны 430 - 450 нм. Источник света в ФЭК-56, ФЭК-56М - лампа накаливания. В кюветы сравнения помещают дистиллированную воду.
Количество шоколада в г на 200 см3 напитка определяют по калибровочному графику или методом сравнения с оптической плотностью контрольного эталонного напитка.
Расчет по калибровочному графику. Для построения калибровочного графика в пяти колбах готовят по рец. 1029 (кол. I) порции по 200 см3, содержащие разное количество шоколада того же сорта, из которого приготовлен исследуемый напиток (табл. 38). Навески измельченного шоколада взвешивают с точностью до 0,01 г.
Определение количества шоколада в напитке "шоколад" по эталонам
|
№ колбы |
Шоколад, г |
Сахар, г |
Молоко, см3 |
Вода, см3 |
Оптическая плотность (ФЭК-56М, светофильтр № 4) |
|
1 |
4,00 |
30 |
180 |
38 |
0,180 |
|
2 |
6,00 |
30 |
180 |
36 |
0,260 |
|
3 |
8,00 |
30 |
180 |
34 |
0,340 |
|
4 |
10,00 |
30 |
180 |
32 |
0,435 |
|
5 |
12,00 |
30 |
180 |
30 |
0,510 |
Каждый эталон анализируют, измеряют оптическую плотность, значения которой записывают в таблицу. Строят калибровочный график: на оси абсцисс указывают содержание шоколада на 200 см3 напитка (С, г), на оси ординат - значения оптической плотности. Пример калибровочного графика, построенного по данным табл. 38, указан на рис. 4.
Рис. 4. Калибровочный график
Расчет методом сравнения с оптической плотностью контрольного напитка. В соответствии с рец. 1029 готовят контрольный напиток (200 см3), содержащий 12,00 г шоколада того же сорта, из которого приготовлен исследуемый напиток. Оба напитка, исследуемый и контрольный, анализируют параллельно по приведенной ранее методике. Количество шоколада (Х, г) в 200 см3 исследуемого напитка находят по формуле
|
|
(90) |
где:
С - известная концентрация вещества в контрольном растворе;
Дх и Дк - величины оптической плотности соответственно исследуемого и контрольного растворов.
Пример расчета. На анализ доставлен напиток "Шоколад" (200 см3), приготовленный по рец. 1029, кол. I. По этой же рецептуре готовят 200 см3 контрольного напитка: шоколад - 12 г, сахар - 30 г, молоко - 180 см3, вода - 30 см3. Результаты измерения: оптическая плотность исследуемого напитка (Дх) = 0,485 , контрольного (Дк) = 0,510.
Количество шоколада (Х, г) в порции 200 см3 исследуемого напитка равно
![]()
Заключение. Исследуемый напиток соответствует рецептуре. Разность 0,59 (12,00 - 11,41) не превышает допустимого 10-процентного предела отклонений, которые при закладке 12 г составляют ±1,20 г.
4.6.4. Кипяченое молоко
При кипячении молока его состав изменяется незначительно: на 0,001 - 0,005 % увеличивается плотность (за счет удаления влаги), на 0,1 - 0,3 % возрастает количество лактозы. Поэтому возможное разбавление кипяченого молока водой можно контролировать по плотности и содержанию лактозы.
4.6.4.1. Плотность молока
Для испытания отбирают по 250 см3 кипяченого и пастеризованного молока. Пробы кипяченого молока хорошо перемешивают и фильтруют через двойной слой марли. В мерный цилиндр вместимостью 250 см3 наливают молоко и медленно опускают в него лактоденсиметр, который не должен касаться стенок и дна цилиндра. Через 2 мин после того, как лактоденсиметр станет неподвижным, производят отсчет показаний температуры и плотности. Отсчет плотности производят по верхнему краю мениска с точностью до 0,0005, отсчет температуры - с точностью до 0,5 °C (ГОСТ 3625-84).
Расхождения между параллельными определениями плотности молока должны быть не более 0,0005. Если температура молока при измерении плотности была выше или ниже 20 °C, то, пользуясь табл. 39, показания лактоденсиметра приводят к температуре 20 °C.
В табл. 39 плотность молока выражена в градусах лактоденсиметра, которые являются дробной частью плотности, увеличенной в тысячу раз. Например, плотность молока 1,026 соответствует 26 градусам лактоденсиметра.
При использовании табл. 39 данные отсчета переводят в градусы лактоденсиметра.
Пересчет плотности коровьего молока
|
Плотность по отсчету лактоденсиметра, град. |
Температура молока, °С |
||||||||||
|
15 |
16 |
17 |
18 |
19 |
20 |
21 |
22 |
23 |
24 |
25 |
|
|
Плотность в град. лактоденсиметра, приведенная к 20 °C |
|||||||||||
|
25 |
24,0 |
24,2 |
24,4 |
24,6 |
24,8 |
25,0 |
25,2 |
25,4 |
25,6 |
25,8 |
26,0 |
|
26 |
25,0 |
25,2 |
25,4 |
25,6 |
25,8 |
26,0 |
26,2 |
26,4 |
26,6 |
26,8 |
27,0 |
|
27 |
25,9 |
26,1 |
26,3 |
26,5 |
26,8 |
27,0 |
27,2 |
27,5 |
27,7 |
27,9 |
28,1 |
|
28 |
26,8 |
27,0 |
27,3 |
27,5 |
27,8 |
28,0 |
28,2 |
28,5 |
28,7 |
29,0 |
29,2 |
|
29 |
27,8 |
28,0 |
28,3 |
28,5 |
28,8 |
29,0 |
29,2 |
29,5 |
29,7 |
30,0 |
30,2 |
|
30 |
28,8 |
29,0 |
29,3 |
29,5 |
29,8 |
30,0 |
30,2 |
30,5 |
30,7 |
31,0 |
31,2 |
|
31 |
29,8 |
30,1 |
30,3 |
30,5 |
30,8 |
31,0 |
31,2 |
31,5 |
31,7 |
32,0 |
32,2 |
|
32 |
30,7 |
31,0 |
31,2 |
31,5 |
31,8 |
32,0 |
32,3 |
32,5 |
32,8 |
33,0 |
33,3 |
|
33 |
31,7 |
32,0 |
32,2 |
32,5 |
32,8 |
33,0 |
33,3 |
33,5 |
33,8 |
34,1 |
34,3 |
|
34 |
32,7 |
33,0 |
33,2 |
33,5 |
33,8 |
34,0 |
34,2 |
34,4 |
34,8 |
35,1 |
35,3 |
|
35 |
33,7 |
34,0 |
34,2 |
34,4 |
34,7 |
35,0 |
35,3 |
35,5 |
35,8 |
36,1 |
36,3 |
|
36 |
34,7 |
34,9 |
35,2 |
35,6 |
35,7 |
36,0 |
36,2 |
36,5 |
36,7 |
37,0 |
36,3 |
Если плотность кипяченого молока ниже, чем пастеризованного, значит, кипяченое молоко разбавлено водой.
Лактозу определяют ускоренным цианидным методом. Массовая доля ее в кипяченом молоке должна быть выше, чем в пастеризованном.
4.6.5. Плодово-ягодные прохладительные напитки, соки
Качество плодово-ягодных прохладительных напитков собственного производства контролируют по массовой доле сухих веществ, определяемых рефрактометрически.
Полученные данные сравнивают с массовой долей сухих веществ контрольного напитка, приготовленного по рецептуре.
При реализации соков промышленного производства разбавление их водой можно установить по относительной плотности.
Относительная плотность соков без мякоти (натуральных и неосветленных, а также с сахаром) по мере разведения их питьевой водой изменяется линейно. В координатах "количество добавленной воды - относительная плотность" зависимость выражается прямой линией, угол наклона которой зависит от содержания воды в соке.
4.6.5.1. Определение относительной плотности соков
Метод основан на том, что ареометр, погруженный в жидкость, опускается до тех пор, пока масса вытесненной им жидкости не будет равна массе ареометра. По глубине погружения, которую показывает шкала ареометра, определяют плотность испытуемой жидкости.
Аппаратура, материалы. Денсиметр или ареометр стеклянный, градуированный при 20 °C; цилиндр мерный; термометр лабораторный с диапазоном измерения 100 °C, ценой деления 1 °C.
Проведение испытания. В сухой стеклянный цилиндр, диаметр которого в 2 - 3 раза больше диаметра утолщенной части ареометра, осторожно (чтобы не образовалась пена) переливают порцию исследуемого сока без мякоти (натурального осветленного или неосветленного, а также с сахаром), температура которого должна быть 20 °C. Определяют объем порции, после чего в жидкость опускают чистый и сухой ареометр таким образом, чтобы не смочить часть прибора, находящегося над жидкостью. Когда ареометр примет устойчивое положение, по нижнему краю мениска снимают показания с точностью до третьего десятичного знака.
При снятии показаний глаз наблюдателя находится на одной горизонтальной плоскости с поверхностью жидкости. Во время определения следят за тем, чтобы ареометр не прикасался к стенкам цилиндра. Затем, вскрыв баллон с соком того же наименования, проверяют плотность контрольного образца сока. Если плотность проверяемого сока меньше плотности контрольного сока, объем воды, добавленный к порции сока, определяют по формуле
|
|
(91) |
где:
V - объем воды, добавленной в порцию сока, см3;
Vск - объем порции исследуемого сока, см3;
Дск - относительная плотность исследуемого сока;
Дк - относительная плотность контрольного сока.
Определение относительной плотности соков с мякотью следует производить сразу же после тщательного взбалтывания их, так как по мере оседания мякоти плотность изменяется. Плотность соков с мякотью по мере разведения их водой изменяется нелинейно.
4.6.5.2. Определение концентрации (степени разбавления) плодовых, ягодных и овощных соков
Анализ в видимой области спектра
Метод основан на цветной реакции природных полифенольных соединений соков с диазосолями. Интенсивность образующейся оранжевой или красной окраски линейно зависит от концентрации сока.
Аппаратура, материалы, реактивы. Фотоэлектроколориметр, обеспечивающий измерения в интервалах волн 315 - 670 нм; весы лабораторные; пипетки вместимостью 2 см3, 5 см3, 10 см3 с делениями; колбы мерные вместимостью 250 см3; пробирки химические; бумага фильтровальная, медленно фильтрующая "синяя лента"; натрия карбонат, раствор с массовой долей 15 %; серная кислота с массовой долей 5 % (20 см3 концентрированной серной кислоты осторожно вливают в 940 см3 дистиллированной воды); пара-нитроанилин (4-нитроанилин), раствор с массовой долей 0,5 % в 5-процентной серной кислоте: 0,5 г пара-нитроанилина растворяют при слабом нагревании в 99,5 см3 раствора серной кислоты, охлаждают до комнатной температуры, фильтруют (вместо пара-нитроанилина можно использовать пара-аминобензойную кислоту, раствор готовят аналогично); нитрат натрия, раствор с массовой долей 0,5 % (0,5 г нитрата натрия растворяют в 99,5 см3 дистиллированной воды, хранят не более 10 сут в плотно укупоренной склянке из оранжевого стекла); раствор спирта в воде в соотношении примерно по объему 1:1 (50 см3 этилового спирта 96° с 50 см3 дистиллированной воды).
Подготовка к испытанию. Раствор диазореактива свежеприготовленный: в день проведения анализа один объем раствора пара-нитроанилина (или пара-аминобензойной кислоты) с массовой долей 0,5 % в растворе серной кислоты с массовой долей 5 % смешивают с двумя объемами раствора нитрата натрия с массовой долей 0,5 %. Реактив готов к применению через 2 - 3 мин и годен в течение 1 дня (диазореактив из пара-аминобензойной кислоты годен только в течение 3 ч).
Проведение испытания. В мерную колбу вместимостью 100 см3 вносят пипеткой 10 см3 анализируемого сока (или взвешивают 10 г сока с мякотью), приливают до метки дистиллированную воду, энергично перемешивают 20 - 30 с и фильтруют через сухой бумажный фильтр (бумага "синяя лента") в сухую коническую колбу. Отбирают пипеткой 5 см3 фильтрата в сухую пробирку и приливают пипетками последовательно 5 см3 этилового спирта, 2 см3 раствора соды и 2 см3 свежеприготовленного диазореактива. Перемешивают и точно через 3 мин измеряют оптическую плотность на фотоэлектроколориметре при длине волны 540 нм в кюветах 20 мм. В кюветы сравнения наливают дистиллированную воду. Если измеренная величина превышает 1,2, то переходят на работу с кюветами 10 мм.
Концентрацию, % сока находят по калибровочному графику, который строят следующим образом. Вскрывают баллон с соком того же вида и той же партии выработки, что и исследуемый сок. Готовят пять калибровочных разведений: в пять мерных колб вместимостью каждая 100 см3 отбирают пипеткой 2, 4, 6, 8, 10 см3 (или г)*(25) сока из вскрытого баллона, доводят объем дистиллированной водой до метки, перемешивают, получая соответственно растворы с концентрацией сока 20, 40, 60, 80, 100 %. Полученные растворы фильтруют через двойной фильтр из бумаги "синяя лента" и анализируют по приведенной выше методике с диазореактивом. По результатам измерения оптической плотности строят калибровочный график: ось абсцисс - % концентрации сока (С), ось ординат - (Д) оптическая плотность (график строят на миллиметровой бумаге).
________________
*(25) Для соков с мякотью берут навески на химических весах.
Анализ в ультрафиолетовой области спектра
Метод основан на том, что природные полифенолы плодов, ягод и овощей поглощают УФ-лучи в области 310 - 370 нм; интенсивность поглощения пропорциональна концентрации сока. Сахар и пищевые кислоты не поглощают лучи в указанной области спектра и не мешают анализу.
Измерение оптической плотности проводят на фотоэлектроколориметрах при 315 нм или при 364 нм (светофильтр № 1 или № 2).
Аппаратура, материалы. Фотоэлектроколориметр, обеспечивающий измерения в интервалах волн 315 - 670 нм; весы лабораторные; пипетки вместимостью 2 см3, 5 см3, 10 см3 с делениями; колбы мерные вместимостью 100 см3; воронки стеклянные; бумага фильтровальная, медленно фильтрующая "синяя лента".Проведение испытания. Пробу сока (5 см3, или 5,00 г) переносят в мерную колбу вместимостью 100 см3, доводят объем до метки дистиллированной водой, энергично перемешивают, фильтруют через двойной фильтр "синяя лента" и измеряют оптическую плотность при 315 нм или 364 нм в кювете 20 мм, в кюветах сравнения - дистиллированная вода. Если величина Д превышает 1,2 - переходят на работу с кюветами 10 мм.
Концентрацию исследуемого сока устанавливают по калибровочному графику. График строят на основе измерения оптической плотности при 315 нм или при 364 нм пяти калибровочных разведений сока из вновь откупоренного баллона. Приготовление калибровочных разведений сока описано выше.
4.6.6. Коктейли с молочными продуктами
Соблюдение рецептуры коктейлей с молочными продуктами контролируют по содержанию жира и сухих веществ. Перед испытанием коктейли перемешивают.
Массовую долю жира в молочных коктейлях определяют методом Гербера (см. с. 31).
Массовую долю сухих веществ определяют высушиванием в сушильном шкафу (ГОСТ 3626-73, раздел 4) (см. с. 8).
4.6.6.1. Расчет правильности соблюдения рецептуры коктейлей с молочными продуктами
В рецептуру коктейлей с молочными продуктами входят продукты, которые отмеривают по объему (сливки, молоко, сиропы, соки) или взвешивают (мороженое, мед и др.). Для проверки правильности соблюдения рецептуры выход порции приводят к единым единицам измерения - граммам. Для этого количество жидких компонентов коктейлей пересчитывают в граммы с учетом их относительной плотности (табл. 40).
Таблица 40
Плотность компонентов коктейлей
|
Продукты |
Плотность (пределы) |
|
Молоко коровье пастеризованное |
1,027 ... 1,03 |
|
Сливки 10-%-ной жирности |
1,017 … 1,018 |
|
Сиропы натуральные |
1,27 ... 1,33 |
|
Соки плодовые и ягодные натуральные |
1,04 ... 1,06 |
Относительную плотность замеряют, как указано в разделе "Плодово-ягодные прохладительные напитки и соки". Для расчета выхода порции коктейля полученные величины в граммах суммируют.
Расчетную массу сухих веществ и жира в коктейлях (в г) находят суммированием количества сухих веществ и жира в продуктах, входящих в коктейль (по данным таблиц химического состава пищевых продуктов, 1987 г.).
Если при анализе компонентов коктейля установлено, что масса сухих веществ и жира ниже, чем предусмотрено нормативно-технической документацией, расчет ведут с учетом фактической массы сухих веществ и жира.
Потери сухих веществ при изготовлении коктейлей составляют 5 % общего количества сухих веществ (в г), введенных в коктейль с продуктами. Отклонение массы при порционировании допускается в размере ±3 %.
Пример расчета. Коктейль приготовлен по рец. 1058: молоко коровье пастеризованное - 100 см3, мороженое сливочное - 25 г, сироп малиновый - 25 см3. Выход - 150 см3. Выход порции - 160 г (молоко - 103, мороженое - 25, сироп малиновый - 32).
Массовая доля сухих веществ и жира в порции коктейля приведена в табл. 41.
Таблица 41
Массовая доля сухих веществ и жира в одной порции коктейля
|
Продукты |
Количество, г |
Массовая доля |
|||
|
сухих веществ |
жира |
||||
|
% |
г |
% |
г |
||
|
Молоко коровье пастеризованное |
103 |
11,5 |
11,84 |
3,2 |
3,30 |
|
Мороженое сливочное |
25 |
34 |
8,50 |
10,0 |
2,50 |
|
Сироп малиновый |
32 |
62,4 |
19,97 |
- |
- |
|
Масса продуктов |
160 |
- |
40,31 |
- |
5,80 |
Потери сухих веществ при изготовлении коктейля составят 2 г (40,31∙0,05). Минимальное допустимое содержание сухих веществ в порции 38,3 г (40,31 - 2), или 24 %.
Потери жира составят 0,29 г (5,8∙0,05), минимальное
допустимое содержание жира 5,51 г (5,80 - 0,29), или 3,4 % ![]() .
.
4.6.7. Алкогольные напитки
Соблюдение рецептуры при изготовлении алкогольных коктейлей следует контролировать по содержанию этилового спирта и общего экстракта.
4.6.7.1. Содержание этилового спирта в жидкой части коктейля определяют по ГОСТ 13191-73 арбитражным методом с помощью стеклянного спиртомера после предварительной перегонки.
Если объем порции коктейля менее 200 см3, для отгонки этилового спирта отбирают 100 см3 и проводят перегонку. Содержание спирта в дистилляте в объемных процентах определяют по ГОСТ 3639-61 с погрешностью не более 0,01 об. % после разведения его дистиллированной водой в 2,5 раза. Для этого дистиллят после прекращения перегонки переносят из приемной колбы вместимостью 100 см3 в мерную колбу вместимостью 250 см3, обмывая несколько раз приемную колбу водой, не доводя объем до метки на 4 - 5 мм. Содержимое колбы энергично перемешивают, колбу плотно закрывают пробкой и оставляют на 30 мин в термостате или водяной бане при температуре 20 °C. Далее содержимое колбы доводят до метки дистиллированной водой, хорошо перемешивают и переливают в сухой стеклянный цилиндр вместимостью 250 см3. Разведение дистиллята затем учитывают при расчете крепости коктейля.
Допустимые расхождения между параллельными определениями не должны превышать 0,06 об. %. За окончательный результат анализа принимают среднее арифметическое двух параллельных определений, вычисленное с точностью до 0,1 об. %.
Обработка результатов. Содержание этилового спирта в коктейле (Х, об. %) рассчитывают по формуле
|
X = а∙в∙к, |
(92) |
где:
a - содержание этилового спирта в отгоне, об. %;
в - кратность разведения отгона;
к - соотношение фактического объема порции коктейля и выхода по рецептуре.
Результат анализа сравнивают с минимально допустимым расчетным значением по рецептуре или с данными анализа контрольного образца (эталона).
Пример расчета. Объем доставленной порции коктейля
"Фантазия": Vфакт = 107 см3
(по рец. 116 ± 3 см3); показания спиртомера a = 16,1 об. %;
для отгона спирта взято 100 см3; объем отгона после разведения - 250
см3. Следовательно, ![]() .
.
Находим содержание этилового спирта в порции коктейля в процентах
![]()
Находим содержание этилового спирта в порции коктейля в см3, если в 100 см3 содержится 40,25 см3 (16,1∙2,5):
|
100 - 40,25 см3 |
|
|
|
107 - X |
Для коктейля "Фантазия" минимально допустимое расчетное содержание спирта - 36,3 об. %, или 42,2 см3, максимальное - 37,5 об. %, или 43,5 см3 (см. пример расчета коктейлей выше).
Заключение. Содержание этилового спирта в коктейле в норме, недолив составляет 6 см3 (113 - 107).
4.6.7.2. Определение содержания общего экстракта в алкогольных коктейлях
Метод основан на определении (с помощью рефрактометра) содержания сухих веществ водного раствора экстракта, оставшегося в колбе после перегонки этилового спирта, с последующим определением содержания общего экстракта по таблице.
Аппаратура, материалы. Рефрактометр типа РПЛ-3, УРЛ, РДУ; термостат или водяная баня; термометр ртутный стеклянный лабораторный со шкалой до 100 °C, с ценой деления 0,1 °C; колбы мерные вместимостью 100 и 200 см3; палочка стеклянная; вода дистиллированная.
Проведение испытания. Остаток в колбе после перегонки для определения содержания спирта в коктейлях переносят без потерь дистиллированной водой в мерную колбу (которой отмеривали исследуемый коктейль) вместимостью 100 или 200 см3, не доводя объем до метки на 5 - 6 мм. Колбу выдерживают 20 - 30 мин на водяной бане при 20 °C, а затем ее содержимое доводят дистиллированной водой до метки и тщательно перемешивают.
На поверхность измерительной призмы рефрактометра оплавленной стеклянной палочкой наносят 1 - 2 капли исследуемой жидкости, закрывают камеру верхней призмой и отмечают показания по шкале рефрактометра.
Если показания рефрактометра снимают при температуре выше или ниже 20 °C, вводят температурную поправку (табл. 3). Рефрактометрию проводят 2 - 3 раза, каждый раз применяя новые порции раствора. Расхождение между повторными определениями не должно превышать 0,2 %. За результат принимают среднеарифметическое значение.
Затем по показанию рефрактометра (см. табл. 43) находят содержание общего экстракта в граммах на 100 см3 напитка. Это значение сравнивают с содержанием общего экстракта, полученного в контрольном образце (эталоне) после отгонки этилового спирта.
Допустимое отклонение от данных анализа контрольного образца (эталона) ±0,8 г/100 см3.
Пример расчета. Объем доставленной на анализ порции коктейля "Фантазия" - 107 см3, по рецептуре - 116 ± 3 см3; для отгонки спирта взято 100 см3 коктейля; после отгонки спирта остаток (экстракт) из перегонной колбы перенесен с помощью воды в мерную колбу на 100 см3. Анализом установлено содержание сухих веществ водного раствора экстракта 10,2 и 10,4 % при 22 °C. Среднее значение - 10,3 %. По табл. 3 находим температурную поправку. Она равна 0,14; 10,3 + 0,14 = 10,17 %. Этому значению в табл. 43 соответствует 10,597 или 10,6 г общего экстракта в 100 см3.
Анализом контрольного образца (эталона) определено содержание общего экстракта в коктейле 12 г/100 см3. Следовательно, минимально допустимое содержание общего экстракта - 11,2 г/100 см3.
Заключение. Содержание общего экстракта в коктейле "Фантазия" занижено на 0,6 г/100 см3 (11,2 - 10,6). Учитывая фактический выход порции - 107 см3, вместо 116 ± 3 см3 по рецептуре, произведен недолив сиропа компота из черешни.
Таблица 42
Соотношение между относительной плотностью, массовыми и объемными процентами смесей спирта и воды
|
Плотность смеси (20°/20°) |
Содержание спирта в смеси |
Плотность смеси (20°/20°) |
Содержание спирта в смеси |
||||
|
в массовых % |
в объемных % |
граммов в 100 при 20 °C |
в массовых % |
в объемных % |
граммов в 100 при 20 °C |
||
|
1,0000 |
0,00 |
0,00 |
0,00 |
0,9906 |
5,34 |
6,69 |
5,28 |
|
0,9998 |
0,11 |
0,13 |
0,10 |
0,9904 |
5,46 |
6, 84 |
5,40 |
|
6 |
21 |
27 |
21 |
2 |
59 |
7,00 |
52 |
|
4 |
32 |
40 |
32 |
0 |
72 |
16 |
65 |
|
2 |
43 |
54 |
42 |
0,9898 |
5,84 |
7,31 |
5,77 |
|
0 |
53 |
67 |
53 |
6 |
9 |
47 |
90 |
|
0,9988 |
64 |
81 |
64 |
4 |
6,10 |
63 |
6,02 |
|
6 |
75 |
94 |
74 |
2 |
23 |
79 |
15 |
|
4 |
86 |
1,08 |
85 |
0 |
36 |
95 |
28 |
|
2 |
96 |
21 |
96 |
0,9888 |
49 |
8,12 |
40 |
|
0 |
1,07 |
35 |
1,06 |
6 |
62 |
28 |
53 |
|
0,9978 |
18 |
49 |
17 |
4 |
75 |
44 |
66 |
|
6 |
29 |
62 |
28 |
2 |
88 |
60 |
79 |
|
4 |
40 |
76 |
39 |
0 |
7,01 |
76 |
92 |
|
2 |
50 |
90 |
50 |
0,9878 |
15 |
93 |
7,05 |
|
0 |
61 |
2,03 |
60 |
6 |
28 |
9,10 |
81 |
|
0,9968 |
72 |
17 |
71 |
4 |
42 |
26 |
31 |
|
6 |
83 |
31 |
82 |
2 |
55 |
43 |
44 |
|
4 |
94 |
44 |
93 |
0 |
68 |
59 |
57 |
|
2 |
2,05 |
58 |
2,04 |
0,9868 |
82 |
76 |
70 |
|
0 |
16 |
72 |
15 |
6 |
95 |
92 |
83 |
|
0,9958 |
28 |
86 |
26 |
4 |
8,09 |
10,09 |
96 |
|
6 |
39 |
3,00 |
37 |
2 |
22 |
26 |
8,09 |
|
4 |
50 |
15 |
48 |
0 |
36 |
42 |
22 |
|
2 |
61 |
29 |
59 |
0,9858 |
49 |
59 |
36 |
|
0 |
72 |
43 |
70 |
6 |
53 |
76 |
49 |
|
0,9948 |
84 |
57 |
82 |
4 |
76 |
92 |
62 |
|
6 |
95 |
71 |
93 |
2 |
90 |
11,09 |
75 |
|
4 |
3,06 |
85 |
3,04 |
0 |
9,03 |
26 |
88 |
|
2 |
18 |
4,00 |
16 |
0,9848 |
17 |
43 |
9,02 |
|
0 |
30 |
14 |
27 |
6 |
31 |
60 |
15 |
|
0,9938 |
41 |
29 |
38 |
4 |
45 |
77 |
29 |
|
6 |
53 |
43 |
50 |
2 |
59 |
94 |
42 |
|
4 |
64 |
58 |
61 |
0 |
73 |
12,11 |
56 |
|
2 |
76 |
72 |
73 |
0,9838 |
87 |
28 |
69 |
|
0 |
88 |
87 |
84 |
6 |
10,11 |
45 |
82 |
|
0,9928 |
3,99 |
5,01 |
3,96 |
0,9834 |
10,15 |
12,62 |
9,96 |
|
6 |
4,12 |
16 |
4,08 |
2 |
29 |
80 |
10,10 |
|
4 |
24 |
32 |
20 |
0 |
44 |
97 |
24 |
|
2 |
36 |
47 |
31 |
0,9828 |
58 |
13,15 |
38 |
|
0 |
48 |
62 |
43 |
6 |
72 |
32 |
52 |
|
0,9918 |
60 |
77 |
55 |
4 |
87 |
50 |
65 |
|
6 |
72 |
92 |
67 |
2 |
11,01 |
67 |
79 |
|
4 |
84 |
6,07 |
79 |
0 |
15 |
85 |
93 |
|
2 |
96 |
22 |
91 |
0,9818 |
30 |
14,03 |
11,07 |
|
0 |
5,09 |
38 |
5,03 |
6 |
44 |
21 |
21 |
|
0,9908 |
21 |
53 |
16 |
4 |
59 |
39 |
35 |
|
0,9812 |
11,74 |
14,56 |
11,49 |
0,9714 |
19,22 |
23,61 |
18,63 |
|
0 |
88 |
74 |
64 |
2 |
37 |
79 |
78 |
|
0,9808 |
12,03 |
92 |
78 |
0 |
52 |
97 |
92 |
|
6 |
18 |
15,10 |
92 |
0,9708 |
67 |
24,15 |
19,06 |
|
4 |
32 |
28 |
12,06 |
6 |
82 |
33 |
20 |
|
2 |
47 |
46 |
20 |
4 |
97 |
51 |
34 |
|
0 |
62 |
64 |
34 |
2 |
20,12 |
69 |
48 |
|
0,9798 |
76 |
82 |
48 |
0 |
27 |
86 |
62 |
|
6 |
91 |
16,00 |
62 |
0,9698 |
41 |
25,04 |
76 |
|
4 |
13,06 |
18 |
77 |
6 |
56 |
22 |
90 |
|
2 |
21 |
36 |
91 |
4 |
71 |
39 |
20,04 |
|
0 |
36 |
55 |
13,06 |
2 |
86 |
57 |
18 |
|
0,9788 |
52 |
73 |
20 |
0 |
21,01 |
74 |
32 |
|
6 |
67 |
91 |
35 |
0,9688 |
15 |
92 |
45 |
|
4 |
82 |
17,10 |
49 |
6 |
30 |
26,09 |
59 |
|
2 |
97 |
28 |
64 |
4 |
44 |
26 |
73 |
|
0 |
14,12 |
47 |
79 |
2 |
59 |
43 |
86 |
|
0,9778 |
28 |
66 |
94 |
0 |
73 |
60 |
21,00 |
|
6 |
43 |
85 |
14,08 |
0,9678 |
88 |
78 |
14 |
|
4 |
59 |
18,03 |
23 |
6 |
22,02 |
95 |
27 |
|
2 |
74 |
22 |
38 |
4 |
16 |
27,12 |
40 |
|
0 |
90 |
41 |
53 |
2 |
31 |
29 |
54 |
|
0,9768 |
15,06 |
18,60 |
14,68 |
0,9670 |
22,45 |
27,46 |
21,67 |
|
6 |
21 |
79 |
83 |
0,9668 |
60 |
63 |
81 |
|
4 |
37 |
98 |
98 |
6 |
74 |
80 |
94 |
|
2 |
53 |
19,17 |
15,13 |
4 |
88 |
97 |
22,08 |
|
0 |
68 |
36 |
28 |
2 |
23,03 |
28,14 |
21 |
|
0,9758 |
84 |
55 |
43 |
|
|
|
|
|
6 |
16,00 |
74 |
58 |
0 |
17 |
31 |
34 |
|
4 |
16 |
93 |
73 |
0,9658 |
31 |
47 |
47 |
|
2 |
31 |
20,12 |
88 |
6 |
45 |
64 |
61 |
|
0 |
47 |
31 |
16,03 |
4 |
60 |
81 |
74 |
|
0,9748 |
62 |
50 |
18 |
2 |
74 |
98 |
87 |
|
6 |
78 |
68 |
33 |
0 |
88 |
29,14 |
23,00 |
|
4 |
94 |
87 |
47 |
0,9648 |
24,02 |
31 |
13 |
|
2 |
17,09 |
21,06 |
62 |
6 |
16 |
47 |
26 |
|
0 |
24 |
24 |
76 |
4 |
30 |
64 |
39 |
|
0,9738 |
40 |
42 |
91 |
2 |
44 |
80 |
52 |
|
6 |
55 |
61 |
17,06 |
0 |
58 |
96 |
65 |
|
4 |
70 |
79 |
20 |
0,9638 |
72 |
30,13 |
78 |
|
2 |
85 |
98 |
34 |
6 |
85 |
29 |
91 |
|
0 |
18,01 |
22,16 |
49 |
4 |
99 |
45 |
24,03 |
|
0,9728 |
16 |
34 |
63 |
2 |
25,13 |
61 |
16 |
|
6 |
31 |
52 |
78 |
0 |
26 |
77 |
28 |
|
4 |
46 |
70 |
92 |
0,9628 |
40 |
92 |
41 |
|
2 |
61 |
89 |
18,06 |
6 |
53 |
31,08 |
53 |
|
0 |
76 |
23,07 |
21 |
4 |
66 |
24 |
66 |
|
0,9718 |
92 |
25 |
35 |
2 |
80 |
40 |
78 |
|
6 |
19,07 |
43 |
49 |
0 |
93 |
55 |
90 |
|
0,9618 |
26,06 |
31,71 |
25,02 |
0,9520 |
32,04 |
38,58 |
30,45 |
|
6 |
20 |
86 |
14 |
0,9518 |
15 |
70 |
55 |
|
4 |
33 |
32,01 |
26 |
6 |
26 |
83 |
65 |
|
2 |
46 |
16 |
38 |
4 |
37 |
95 |
75 |
|
0 |
59 |
32 |
50 |
2 |
49 |
39,08 |
85 |
|
0,9608 |
26,72 |
32,47 |
25,62 |
0,9510 |
32,60 |
39,21 |
30,94 |
|
6 |
85 |
62 |
74 |
0,9508 |
71 |
33 |
31,04 |
|
4 |
98 |
77 |
86 |
6 |
82 |
46 |
14 |
|
2 |
27,11 |
92 |
98 |
4 |
93 |
59 |
24 |
|
0 |
24 |
33,07 |
26,10 |
2 |
33,04 |
71 |
34 |
|
0,9598 |
36 |
22 |
22 |
0 |
15 |
84 |
44 |
|
6 |
49 |
36 |
33 |
0,9498 |
26 |
96 |
54 |
|
4 |
62 |
51 |
45 |
6 |
38 |
40,08 |
64 |
|
2 |
74 |
66 |
56 |
4 |
49 |
21 |
73 |
|
0 |
87 |
81 |
68 |
2 |
60 |
33 |
83 |
|
0,9588 |
28,00 |
95 |
80 |
0 |
71 |
46 |
93 |
|
6 |
12 |
34,10 |
91 |
0,9488 |
82 |
58 |
32,03 |
|
4 |
25 |
24 |
27,02 |
6 |
93 |
70 |
13 |
|
2 |
37 |
38 |
14 |
4 |
34,04 |
83 |
22 |
|
0 |
49 |
52 |
25 |
2 |
15 |
95 |
32 |
|
0,9578 |
62 |
66 |
36 |
0 |
26 |
41,07 |
42 |
|
6 |
74 |
81 |
47 |
0,9478 |
36 |
19 |
51 |
|
4 |
86 |
95 |
57 |
6 |
47 |
31 |
61 |
|
2 |
99 |
35,09 |
70 |
4 |
58 |
44 |
70 |
|
0 |
29,11 |
23 |
80 |
2 |
69 |
56 |
80 |
|
0,9568 |
23 |
37 |
91 |
0 |
80 |
68 |
90 |
|
6 |
35 |
50 |
28,02 |
0,9468 |
91 |
80 |
99 |
|
4 |
47 |
64 |
13 |
6 |
35,02 |
92 |
33,09 |
|
2 |
59 |
78 |
24 |
4 |
12 |
42,04 |
18 |
|
0 |
71 |
92 |
35 |
2 |
22 |
16 |
27 |
|
0,9558 |
83 |
36,05 |
46 |
0 |
33 |
27 |
36 |
|
6 |
95 |
19 |
56 |
0,9458 |
44 |
39 |
46 |
|
4 |
30,06 |
33 |
67 |
6 |
54 |
51 |
55 |
|
2 |
18 |
46 |
78 |
4 |
65 |
62 |
64 |
|
0 |
30 |
60 |
88 |
2 |
75 |
74 |
73 |
|
0,9548 |
42 |
73 |
99 |
0 |
86 |
86 |
82 |
|
6 |
54 |
86 |
29,10 |
0,9448 |
96 |
97 |
92 |
|
4 |
65 |
37,00 |
20 |
6 |
36,07 |
43,09 |
34,01 |
|
0,9542 |
30,77 |
37,13 |
29,31 |
0,9444 |
36,17 |
43,20 |
34,10 |
|
0 |
89 |
27 |
41 |
2 |
27 |
32 |
19 |
|
0,9538 |
31,01 |
40 |
52 |
0 |
38 |
43 |
28 |
|
6 |
12 |
53 |
62 |
0,9438 |
48 |
55 |
37 |
|
4 |
24 |
66 |
73 |
6 |
58 |
66 |
46 |
|
2 |
35 |
79 |
83 |
4 |
69 |
78 |
55 |
|
0 |
46 |
92 |
93 |
2 |
78 |
89 |
64 |
|
0,9528 |
58 |
38,06 |
30,04 |
0 |
90 |
44,00 |
73 |
|
6 |
70 |
19 |
14 |
0,9428 |
37,00 |
12 |
82 |
|
4 |
81 |
32 |
24 |
6 |
10 |
23 |
91 |
|
2 |
92 |
45 |
34 |
4 |
20 |
34 |
35,00 |
|
0,9422 |
37,31 |
44,46 |
35,09 |
0,9386 |
39,12 |
46,44 |
36,66 |
|
0 |
41 |
57 |
18 |
4 |
22 |
55 |
74 |
|
0,9418 |
51 |
68 |
26 |
2 |
32 |
66 |
83 |
|
6 |
61 |
79 |
35 |
0 |
42 |
77 |
91 |
|
4 |
71 |
90 |
44 |
0,9378 |
52 |
88 |
37,00 |
|
2 |
82 |
45,02 |
53 |
6 |
62 |
98 |
08 |
|
0 |
92 |
13 |
62 |
4 |
72 |
47,09 |
17 |
|
0,9408 |
38,02 |
24 |
70 |
2 |
82 |
20 |
25 |
|
6 |
12 |
35 |
79 |
0 |
92 |
31 |
34 |
|
4 |
22 |
46 |
88 |
0,9368 |
40,02 |
42 |
42 |
|
2 |
32 |
57 |
96 |
6 |
12 |
52 |
51 |
|
0 |
42 |
68 |
36,05 |
4 |
22 |
63 |
59 |
|
0,9398 |
52 |
79 |
14 |
2 |
31 |
73 |
67 |
|
6 |
62 |
90 |
22 |
0 |
41 |
84 |
76 |
|
4 |
72 |
46,01 |
31 |
0,9358 |
51 |
94 |
84 |
|
2 |
82 |
12 |
40 |
6 |
61 |
48,05 |
92 |
|
0 |
92 |
23 |
48 |
4 |
70 |
16 |
38,01 |
Содержание экстрактивных веществ (по показаниям рефрактометра)
|
Показание рефрактометра, мас. % |
Содержание общего экстракта в граммах на 100 см3 |
Показание рефрактометра, мас. % |
Содержание общего экстракта в граммах на 100 см3 |
|
0,0 |
0,000 |
4,8 |
4,881 |
|
1 |
099 |
9 |
985 |
|
2 |
199 |
5,0 |
5,089 |
|
3 |
299 |
1 |
193 |
|
4 |
399 |
2 |
296 |
|
5 |
500 |
3 |
400 |
|
6 |
600 |
4 |
505 |
|
7 |
700 |
5 |
609 |
|
8 |
800 |
6 |
713 |
|
9 |
900 |
7 |
817 |
|
1,0 |
1,000 |
8 |
922 |
|
1 |
102 |
9 |
6,026 |
|
2 |
203 |
6,0 |
131 |
|
3 |
304 |
1 |
235 |
|
4 |
405 |
2 |
340 |
|
5 |
506 |
3 |
445 |
|
6 |
607 |
4 |
550 |
|
7 |
708 |
5 |
655 |
|
8 |
809 |
6 |
760 |
|
9 |
910 |
7 |
865 |
|
2,0 |
2,012 |
8 |
970 |
|
1 |
113 |
9 |
7,075 |
|
2 |
214 |
7,0 |
180 |
|
3 |
316 |
1 |
286 |
|
4 |
418 |
2 |
392 |
|
5 |
519 |
3 |
497 |
|
6 |
621 |
4 |
603 |
|
7 |
723 |
5 |
709 |
|
8 |
825 |
6 |
815 |
|
9 |
927 |
7 |
921 |
|
3,0 |
3,028 |
7,8 |
8,027 |
|
1 |
132 |
9 |
133 |
|
2 |
234 |
8,0 |
239 |
|
3 |
336 |
1 |
345 |
|
4 |
439 |
2 |
452 |
|
5 |
541 |
3 |
553 |
|
6 |
644 |
4 |
665 |
|
7 |
769 |
5 |
771 |
|
8 |
849 |
6 |
878 |
|
9 |
952 |
7 |
985 |
|
4,0 |
4,055 |
8 |
9,092 |
|
1 |
158 |
9 |
199 |
|
2 |
261 |
9,0 |
306 |
|
3 |
364 |
1 |
413 |
|
4 |
468 |
2 |
520 |
|
5 |
571 |
3 |
627 |
|
6 |
674 |
4 |
735 |
|
7 |
778 |
5 |
832 |
|
9,6 |
9,950 |
14,7 |
15,551 |
|
7 |
10,057 |
8 |
663 |
|
8 |
165 |
9 |
775 |
|
9 |
273 |
15,0 |
887 |
|
10,0 |
381 |
1 |
999 |
|
1 |
489 |
2 |
16,112 |
|
2 |
597 |
3 |
225 |
|
3 |
705 |
4 |
338 |
|
4 |
812 |
5 |
449 |
|
5 |
922 |
6 |
563 |
|
6 |
11,030 |
7 |
676 |
|
7 |
139 |
8 |
789 |
|
8 |
247 |
9 |
902 |
|
9 |
356 |
16,0 |
17,016 |
|
11,0 |
465 |
1 |
129 |
|
1 |
574 |
2 |
242 |
|
11,2 |
11,683 |
16,3 |
17,356 |
|
3 |
792 |
4 |
469 |
|
4 |
901 |
5 |
583 |
|
5 |
12,010 |
6 |
696 |
|
6 |
120 |
7 |
810 |
|
7 |
229 |
8 |
924 |
|
8 |
338 |
9 |
18,038 |
|
9 |
448 |
17,0 |
152 |
|
12,0 |
558 |
1 |
267 |
|
1 |
667 |
2 |
381 |
|
2 |
777 |
3 |
495 |
|
3 |
887 |
4 |
610 |
|
4 |
996 |
5 |
724 |
|
5 |
13,106 |
6 |
839 |
|
6 |
217 |
7 |
954 |
|
7 |
327 |
8 |
19,069 |
|
8 |
437 |
9 |
184 |
|
9 |
548 |
18,0 |
299 |
|
13,0 |
658 |
1 |
413 |
|
1 |
769 |
2 |
529 |
|
2 |
879 |
3 |
644 |
|
3 |
991 |
4 |
759 |
|
4 |
14,102 |
5 |
875 |
|
5 |
213 |
6 |
990 |
|
6 |
324 |
7 |
20,106 |
|
7 |
435 |
8 |
222 |
|
8 |
546 |
9 |
338 |
|
9 |
657 |
19,0 |
455 |
|
14,0 |
769 |
1 |
570 |
|
1 |
880 |
2 |
686 |
|
2 |
992 |
3 |
802 |
|
3 |
15,103 |
4 |
919 |
|
4 |
207 |
5 |
21,035 |
|
5 |
327 |
6 |
152 |
|
6 |
439 |
7 |
268 |
|
19,8 |
21,385 |
24,9 |
27,467 |
|
9 |
502 |
25,0 |
589 |
|
20,0 |
619 |
1 |
711 |
|
1 |
736 |
2 |
833 |
|
2 |
853 |
3 |
955 |
|
3 |
970 |
4 |
28,077 |
|
4 |
22,108 |
5 |
199 |
|
5 |
205 |
6 |
322 |
|
6 |
323 |
7 |
444 |
|
7 |
430 |
8 |
567 |
|
8 |
558 |
9 |
689 |
|
9 |
676 |
26,0 |
813 |
|
21,0 |
794 |
1 |
935 |
|
1 |
912 |
2 |
29,058 |
|
2 |
23,029 |
3 |
182 |
|
3 |
148 |
4 |
305 |
|
4 |
266 |
5 |
428 |
|
5 |
385 |
6 |
552 |
|
6 |
503 |
7 |
675 |
|
7 |
622 |
8 |
798 |
|
8 |
740 |
9 |
923 |
|
9 |
859 |
27,0 |
30,046 |
|
22,0 |
978 |
1 |
170 |
|
1 |
24,087 |
2 |
297 |
|
2 |
216 |
3 |
418 |
|
3 |
335 |
4 |
543 |
|
4 |
454 |
5 |
667 |
|
5 |
574 |
6 |
792 |
|
6 |
693 |
7 |
916 |
|
7 |
812 |
8 |
31,041 |
|
8 |
931 |
9 |
165 |
|
9 |
25,052 |
28,0 |
290 |
|
23,0 |
25,172 |
28,1 |
31,415 |
|
1 |
292 |
2 |
540 |
|
2 |
412 |
3 |
665 |
|
3 |
532 |
4 |
791 |
|
4 |
652 |
5 |
916 |
|
5 |
772 |
6 |
32,042 |
|
6 |
893 |
7 |
167 |
|
7 |
26,013 |
8 |
293 |
|
8 |
134 |
9 |
418 |
|
9 |
254 |
29,0 |
545 |
|
24,0 |
375 |
1 |
671 |
|
1 |
496 |
2 |
797 |
|
2 |
617 |
3 |
923 |
|
3 |
738 |
4 |
33,049 |
|
4 |
859 |
5 |
176 |
|
5 |
981 |
6 |
302 |
|
6 |
27,102 |
7 |
429 |
|
7 |
224 |
8 |
555 |
|
8 |
345 |
9 |
683 |
|
30,0 |
33,779 |
33,0 |
37,668 |
|
1 |
936 |
1 |
798 |
|
2 |
34,064 |
2 |
928 |
|
3 |
191 |
3 |
38,059 |
|
4 |
318 |
4 |
189 |
|
5 |
456 |
5 |
320 |
|
6 |
574 |
6 |
451 |
|
7 |
701 |
7 |
582 |
|
8 |
829 |
8 |
713 |
|
9 |
957 |
9 |
844 |
|
31,0 |
35,085 |
34,0 |
976 |
|
1 |
216 |
1 |
39,107 |
|
2 |
341 |
2 |
238 |
|
3 |
469 |
3 |
370 |
|
4 |
598 |
4 |
502 |
|
31,5 |
35,726 |
34,5 |
39,634 |
|
6 |
852 |
6 |
766 |
|
7 |
984 |
7 |
898 |
|
8 |
36,113 |
8 |
40,023 |
|
9 |
242 |
9 |
162 |
|
32,0 |
371 |
35,0 |
295 |
|
1 |
500 |
1 |
427 |
|
2 |
629 |
2 |
559 |
|
3 |
759 |
3 |
692 |
|
4 |
888 |
4 |
825 |
|
5 |
37,018 |
5 |
947 |
|
6 |
148 |
6 |
41,091 |
|
7 |
278 |
7 |
244 |
|
8 |
408 |
8 |
357 |
|
9 |
538 |
9 |
507 |
4.6.7.3. Методика составления и расчета рецептур алкогольных коктейлей
В связи с тем, что при изготовлении коктейлей жидкие компоненты, содержащие спирт (вина, водки, коньяки, ром, ликеры, настойки), сиропы, соки, минеральные воды отмеривают по объему специальной посудой, имеющей клеймо госповерки, а твердые компоненты (дольки цитрусовых, консервированные фрукты, орехи, лед и др.) взвешивают, при составлении рецептур коктейлей указанные компоненты необходимо суммировать отдельно в объемных и весовых измерениях и выход порции записывать дробью, в числителе которой - объем жидкой части, в знаменателе - масса плотной части*(26).
_________________
*(26) Количество взвешенного льда учитывают в объеме напитка (1 г = 1 см3)
При составлении рецептур коктейлей с использованием компонентов, кроме консервированных фруктов компота, следует использовать также сироп в таком процентном соотношении, как он содержится в консервах. Например, при составлении рецептуры коктейля использовали 10 г черешни. Согласно ГОСТ 816-81, в этом компоте должно быть плодов не менее 55 % к массе нетто компота. Следовательно, количество сиропа, которое необходимо использовать для коктейля, составит:
|
10 - 55 |
|
|
|
Х - 45 |
Переводим весовые единицы измерения в объемные, учитывая относительную плотность сиропов: р 20°/4° = 1,04 - 1,085 (табл. 44).
Таблица 44
Зависимость между плотностью и процентным содержанием сухих веществ (извлечение из ГОСТ 8756.2-82)
|
Плотность при 20°/4° |
Содержание сухих веществ, мас. % |
Плотность при 20°/4° |
Содержание сухих веществ, мас. % |
Плотность при 20°/4° |
Содержание сухих веществ, мас. % |
|
1,0390 |
10,2 |
1,0541 |
13,8 |
1,0695 |
17,4 |
|
1,0394 |
10,3 |
1,0545 |
13,9 |
1,0700 |
17,5 |
|
1,0398 |
10,4 |
1,0549 |
14,0 |
1,0704 |
17,6 |
|
1,0402 |
10,5 |
1,0553 |
14,1 |
1,0708 |
17,7 |
|
1,0406 |
10,6 |
1,0558 |
14,2 |
1,0713 |
17,8 |
|
1,0410 |
10,7 |
1,0562 |
14,3 |
1,0717 |
17,9 |
|
1,0415 |
10,8 |
1,0566 |
14,4 |
1,0721 |
18,0 |
|
1,0419 |
10,9 |
1,0570 |
14,5 |
1,0726 |
18,1 |
|
1,0423 |
11,0 |
1,0575 |
14,6 |
1,0730 |
18,2 |
|
1,0427 |
11,1 |
1,0579 |
14,7 |
1,0735 |
18,3 |
|
1,0431 |
11,2 |
1,0583 |
14,8 |
1,0739 |
18,4 |
|
1,0435 |
11,3 |
1,0587 |
14,9 |
1,0743 |
18,5 |
|
1,0440 |
11,4 |
1,0592 |
15,0 |
1,0748 |
18,6 |
|
1,0444 |
11,5 |
1,0596 |
15,1 |
1,0752 |
18,7 |
|
1,0448 |
11,6 |
1,0600 |
15,2 |
1,0757 |
18,8 |
|
1,0452 |
11,7 |
1,0605 |
15,3 |
1,0761 |
18,9 |
|
1,0456 |
11,8 |
1,0609 |
15,4 |
1,0766 |
19,0 |
|
1,0460 |
11,9 |
1,0612 |
15,5 |
1,0770 |
19,1 |
|
1,0465 |
12,0 |
1,0617 |
15,6 |
1,0774 |
19,2 |
|
1,0469 |
12,1 |
1,0622 |
15,7 |
1,0779 |
19,3 |
|
1,0473 |
12,2 |
1,0626 |
15,8 |
1,0783 |
19,4 |
|
1,0477 |
12,3 |
1,0630 |
15,9 |
1,0787 |
19,5 |
|
1,0481 |
12,4 |
1,0635 |
16,0 |
1,0792 |
19,6 |
|
1,0486 |
12,5 |
1,0639 |
16,1 |
1,0796 |
19,7 |
|
1,0490 |
12,6 |
1,0643 |
16,2 |
1,0801 |
19,8 |
|
1,0494 |
12,7 |
1,0648 |
16,3 |
1,0805 |
19,9 |
|
1,0498 |
12,8 |
1,0652 |
16,4 |
1,0810 |
20,0 |
|
1,0502 |
12,9 |
1,0656 |
16,5 |
1,0814 |
20,1 |
|
1,0507 |
13,0 |
1,0661 |
16,6 |
1,0818 |
20,2 |
|
1,0511 |
13,1 |
1,0665 |
16,7 |
1,0823 |
20,3 |
|
1,0515 |
13,2 |
1,0669 |
16,8 |
1,0827 |
20,4 |
|
1,0519 |
13,3 |
1,0674 |
16,9 |
1,0832 |
20,5 |
|
1,0524 |
13,4 |
1,0678 |
17,0 |
1,0836 |
20,6 |
|
1,0528 |
13,5 |
1,0682 |
17,1 |
1,0841 |
20,7 |
|
1,0532 |
13,6 |
1,0687 |
17,2 |
1,0845 |
20,8 |
|
1,0536 |
13,7 |
1,0691 |
17,3 |
1,0850 |
20,9 |
Согласно ГОСТ 816-81, содержание сухих веществ в сиропе компота из черешни - 19 %, что соответствует плотности 1,0766. Следовательно, 8,18: 1,0766 = 7,6 см3 или 8 см3 сиропа.
Экспериментально установлено, что безвозвратные потери при изготовлении коктейлей составляют 3 % от выхода по рецептуре (в см3). Кроме того, действующими сборниками рецептур при порционировании допускается отклонение 3 % от выхода как в меньшую, так и в большую сторону.
Из табл. 45 видно, что выход жидкой части порции коктейля составляет 120 см3. Учитывая безвозвратные потери (-3 %), выход порции составит 116 см3. С учетом отклонений при порционировании (+3 %) выход жидкой части порции коктейля будет 116 ± 3 см3. Выход фруктов с учетом допустимых отклонений ±10 % (табл. 45) составит 10 ± 1 г.
Таблица 45
|
Наименование коктейля и его компонентов |
№ стандарта |
Количество |
|
|
см3 |
г |
||
|
"Фантазия"* |
|
|
|
|
Коньяк (3 звездочки) |
ГОСТ 13741-78 |
40 |
- |
|
Ром "Порторико" |
Контракт на импортную продукцию |
30 |
- |
|
Ликер "Шартрез" |
ГОСТ 7190-71 |
25 |
- |
|
Компот "Черешня" |
ГОСТ 816-81 |
- |
- |
|
Сироп |
- |
8 |
- |
|
Фрукты |
- |
- |
10 |
|
Лед пищевой |
- |
17 |
17 |
|
Итого |
|
120/10 |
- |
|
Потери безвозвратные (-3 %) |
|
4 |
- |
|
Выход |
|
116 ± 3 |
10 ± 1 |
_____________
* Рецептура утверждена Главным управлением общественного питания Ленгорисполкома, 1978 г.
Расчет содержания этилового спирта. Содержание этилового спирта в порции коктейля находят суммированием количества спирта в см3, содержащегося в напитках, входящих в коктейль. Для этого содержание безводного этилового спирта каждого напитка, входящего в состав коктейля, выраженное в объемных процентах (градусах крепости -1° = 1 % объемному), находят в нормативно-технической документации и пересчитывают его на количество каждого компонента, входящего в рецептуру*(27).
_______________
*(27) Если содержание этилового спирта в ГОСТах (для вин шампанских, виноградных марочных и крепленых) дано в интервале 10 - 13,5, 10 - 12 об. %, для расчета рецептуры принимают среднее значение; если в ГОСТах показатель содержания спирта дан на группу вин или ликеров (ГОСТ 17292-83, ГОСТ 7190-71 и др.), для расчета рецептуры используют крепость, указанную на этикетке бутылки заводской упаковки.
Пример расчета. В коктейль "Фантазия" входят 3 компонента, содержащие спирт: коньяк, ром и ликер.
Согласно ГОСТ 13741-78 в
коньяке (3 звездочки) содержание спирта (крепость) - 40°, или 40 об. %. В
данный коктейль входит 40 см3 коньяка. В 40 см3 коньяка
содержится 16 см3 ![]() безводного этилового спирта. Такой же
расчет производят по другим компонентам, входящим в состав коктейля, и получают
суммарное содержание спирта (табл. 46).
безводного этилового спирта. Такой же
расчет производят по другим компонентам, входящим в состав коктейля, и получают
суммарное содержание спирта (табл. 46).
Таблица 46
|
Наименование коктейля и его компонентов |
Стандарт на продукцию |
см3 |
г |
Спирт этиловый безводный |
|
|
об.% |
см3 |
||||
|
"Фантазия" |
|
|
|
|
|
|
Коньяк(3 звездочки) |
ГОСТ 13741-78 |
40 |
- |
40 |
16 |
|
Ром "Порторико" |
Контракт на импортную продукцию |
30 |
|
60 |
18 |
|
Ликер "Шартрез" |
ГОСТ 7190-71 |
25 |
- |
44 |
11 |
|
Компот "Черешня" |
ГОСТ 816-81 |
- |
- |
- |
- |
|
Сироп |
- |
8 |
- |
- |
- |
|
Фрукты |
- |
- |
10 |
- |
- |
|
Лед пищевой |
- |
17 |
17 |
- |
- |
|
Итого |
|
120/10 |
- |
- |
45 |
|
Потери безвозвратные (-3 %) |
|
4 |
|
|
|
|
Выход |
|
116 ± 3 |
|
|
|
Из табл. 46 видно, что если в 120 см3 жидкой части коктейля содержится 45 см3 безводного этилового спирта, то в 116 см3 содержится 43,5 см3 спирта. Учитывая допуск - 3 %, рассчитывают минимально допустимое содержание спирта в 116 см3 коктейля: 3 % от 43,5 см3 составит 1,3 см3. Следовательно, минимально допустимое содержание спирта в порции 43,5 - 1,3 = 42,2 см3, максимальное - 43,5 см3.
Далее рассчитывают минимально допустимое содержание спирта в объемных процентах, исходя из пропорции
|
116 - 42,2 |
|
|
|
100 - Х |
Следовательно, содержание спирта в коктейле "Фантазия" должно быть не менее 36,3 об. %.
Максимально допустимое содержание спирта в коктейле
![]()
В случае отклонения фактического выхода порции коктейля от выхода по рецептуре (в кубических сантиметрах) результат анализа по содержанию спирта (в процентах) умножают на коэффициент K, указывающий соотношение фактического объема и объема порции по рецептуре
|
|
(93) |
Раздел I, Часть II
4.7. Мучные кулинарные изделия
Мучные кулинарные изделия изготавливают по нормативно-технической документации, в соответствии с которой их и контролируют, или по Сборникам рецептур. Изделия, приготовляемые по Сборникам, исследуют по аналогии с теми, на которые имеется нормативно-техническая документация.
4.7.1. Пирожки печеные и жареные из дрожжевого теста (столовые и сдобные)*(1)
Отбор проб, подготовка их к испытаниям, исследуемые физико-химические показатели указаны в прил. 2.
_____________
*(1) По аналогичным показателям с использованием тех же методов контролируют ватрушки сдобные с творогом (ГОСТ 24557-89), приготовляемые по Сборнику рецептур блюд и кулинарных изделий. М., 1981 г., пирожки жареные и печеные из дрожжевого теста, пирожки печеные из слоеного теста, беляши, чебуреки, манты, самсы (РСТ КазССР 917-91), кулинарные и колбасные изделия, запеченные в тесте.
4.7.1.1. Массовая доля фарша (колбасы, сосисок, котлет)
Отобранные изделия взвешивают с точностью до 1 г, разрезают вдоль или на четыре части (вдоль и поперек) и отделяют фарш скальпелем вместе с полужидкой частью основы и основу взвешивают. Массовую долю фарша к массе изделия (X, %) определяют по формуле:
где:
m - масса фарша, г;
m1 - масса изделия, г.
Влажность основы. Основу после отделения фарша измельчают вместе с коркой острым ножом или на мясорубке с мелкой решеткой и определяют массовую долю влаги методом высушивания, как указано на с. 11.
Кислотность основы. Определение проводят арбитражным или ускоренным методом, как описано на с. 78.
Массовую долю жира в основе определяют экстракционно-весовым (ГОСТ 5668-78) или рефрактометрическим методом (см. с. 25, 39).
4.7.1.2. Массовая доля сахара в основе
Определение сахаров проводят перманганатным методом после гидролиза сахарозы и выражают содержание сахаров в процентах на сухое вещество.
Аппаратура, материалы, реактивы. Те же, что на с. 148, а также универсальная индикаторная бумага; фенолфталеин, спиртовой раствор с массовой долей 1 %.
Проведение испытания. 20 г измельченной пробы (для пирожков с фруктовыми начинками - 10 г) переносят с помощью 100 - 125 см3 теплой дистиллированной воды в мерную колбу вместимостью 200 или 250 см3. Органические кислоты, содержащиеся в основе пирожков с фруктовыми фаршами, творогом, морковью с творогом, морковью и яблоками, квашеной капустой и т.д., нейтрализуют раствором карбоната натрия с массовой долей 15 % до pH 7,0 по универсальной индикаторной бумаге. Колбу оставляют на 15 мин, периодически встряхивая. Для осаждения несахаров приливают 15 см3 раствора сульфата цинка массовой концентрации 150 г/дм3 и такое количество раствора гидроокиси натрия массовой концентрации 40 г/дм3, какое установлено предварительным титрованием в присутствии фенолфталеина. Содержимое колбы энергично встряхивают в течение 3 мин, доводят дистиллированной водой до метки, перемешивают, дают 10 - 15 мин, отстояться, затем фильтруют в сухую колбу (первые порции фильтрата отбрасывают).
Гидролиз сахарозы проводят, как описано на с. 148. Массовую долю сахара определяют перманганатным методом по Бертрану (см. с. 47).
При исследовании основы пирожков с фруктовыми фаршами определяют сахара до и после гидролиза тем же методом.
Массовая доля сахара в фарше. Определение сахара в фарше проводят в спорных случаях. Подготовку вытяжки из плодового и плодово-ягодного фарша проводят, как в киселях (см. с. 169), из творожного - как в изделиях из творога (см. с. 165).
Массовая доля сухих веществ в фарше. Массовую долю сухих веществ в фаршах мясном с луком, мясном с рисом определяют в соответствии с ГОСТ 4288-76, фарше творожном - ГОСТ 3626-73, в остальных - ГОСТ 8756.2-82. Масса навески и режим сушки указаны в табл. 2.
Жир фритюрный. Качество фритюрного жира определяют методами, изложенными ниже.
4.7.2. Пироги полуоткрытые дрожжевые с разными фаршами, реализуемые по массе и штучно
Массовая доля фарша. Для определения массовой доли фарша берут целый штучный пирог массой до 1 кг, половину штучного пирога массой 1 кг и более или четверть пирога, реализуемого по массе. Подготовленную пробу пирога взвешивают, отделяют начинку от основы с помощью скальпеля и взвешивают основу. Массовую долю фарша рассчитывают по формуле (94).
Для определения физико-химических показателей: влажности, массовой доли сахара и жира в мякише изделий после удаления фарша срезают корочки у основы (толщиной около 1 см) и мякиш измельчают. Фарш тщательно растирают в ступке.
Влажность, кислотность, массовую долю сахара и жира в основе, массовую долю сухих веществ в фарше определяют так же, как и для пирожков (см. с. 214).
4.7.3. Чебуреки, беляши, манты, самсы
Контроль качества указанных изделий проводят в соответствии с РСТ Казахстана 917-91 по показателям: массовая доля фарша к массе изделия, массовая доля влаги в основе, массовая доля сухих веществ в фарше, массовая доля лука в фарше.
Для лабораторного контроля из средней пробы отбирают 10 шт. мант, по 5 шт. чебуреков и беляшей.
Влажность основы. Массовую долю влаги в основе определяют методом высушивания. Навеску высушивают при температуре 130 °C в течение 30 мин или при температуре 102 ± 2 °C до постоянной массы (см. с. 11).
Массовую долю фарша, кислотность основы и массовую долю жира в ней определяют, как описано на с. 214.
4.7.3.1. Массовая доля лука в фарше
Определение массовой доли лука в фарше проводят по РСТ Казахстана 917-91. При определении количества лука в фаршах изделий дополнительно производят отбор проб лука репчатого из той же партии, которая использовалась для приготовления фарша. Для этого из средней пробы отбирают 3 - 4 луковицы.
Массовую долю лука в фарше определяют по содержанию углеводов.
Аппаратура, материалы, реактивы. Те же, что на с. 74 - 76, а также йодид калия; сульфат цинка 7-водный; крахмал, раствор с массовой долей 1 %.
Подготовка к испытанию. Определение поправочного коэффициента к раствору гексацианоферрата (III) калия массовой концентрации 10 г/дм3.
В коническую колбу вместимостью 250 см3 вносят из бюретки 50 см3 раствора гексацианоферрата (III) калия, прибавляют 3 г йодида калия и 1,5 г сульфата цинка, взбалтывают и тут же титруют раствором тиосульфата натрия концентрации 0,1 моль/дм3 (0,1 н) в присутствии крахмала в качестве индикатора. Поправку рассчитывают по формуле:
|
|
(95) |
где:
V - объем раствора тиосульфата натрия, пошедший на титрование, см3;
0,03291 - количество гексацианоферрата (III) калия, соответствующее 1 см3 раствора тиосульфата натрия концентрации 0,1 моль/дм3 (0,1 н);
0,5 - количество гексацианоферрата (III) калия, содержащегося в 50 см3 раствора массовой концентрации точно 10 г/дм3, г.
Проведение испытания. Для извлечения углеводов от подготовленной пробы фарша берут навеску массой 45 г (манты, самса) и 60 г (чебуреки, беляши) с точностью до 0,01 г, переносят в фарфоровую ступку, добавляют теплую дистиллированную воду (40 - 50 °C), тщательно растирают и переносят количественно в мерную колбу вместимостью 200 см3, смывая прилипшие частицы. Объем жидкости должен быть примерно равен половине объема мерной колбы. Содержимое колбы выдерживают на водяной бане с температурой 50 - 60 °C при частом взбалтывании в течение 15 - 30 мин. для полного перехода углеводов лука в раствор. По охлаждении добавляют в колбу для осаждения "мешающих несахаров" 15 см3 раствора гексацианоферрата (II) калия массовой концентрации 150 г/дм3 и 15 см3 раствора сульфата цинка массовой концентрации 300 г/дм3. Содержимое колбы тщательно перемешивают и доводят водой до метки. Если жидкость над осадком недостаточно прозрачна, количество осадителей увеличивают в 1,5 - 2 раза. Осадок отстаивают 15 - 20 мин, затем раствор фильтруют через вату или складчатый фильтр в сухую колбу. Фильтрат должен быть прозрачным.
Для инверсии сахарозы в мерную колбу вместимостью 100 см3 вносят 50 см3 полученного фильтрата, добавляют 5 см3 концентрированной соляной кислоты. В колбу опускают термометр и помещают в водяную баню, нагретую до 80 - 85 °C, доводят температуру раствора до 67 - 70 °C в течение 2 - 3 мин. и проводят инверсию точно 5 минут. Затем раствор быстро охлаждают до комнатной температуры, ополаскивают и удаляют термометр, нейтрализуют раствором гидроксида натрия (гидроксида калия) массовой концентрации 150 г/дм3 до желто-оранжевого окрашивания в присутствии метилового оранжевого.
Содержимое колбы доводят дистиллированной водой до метки и тщательно перемешивают. В полученном растворе определяют содержание общего сахара цианидным методом (см. с. 55).
Обработка результатов испытания. Массовую долю лука (X, %) вычисляют по формуле:
|
|
(96) |
где:
K - поправка на раствор гексацианоферрата (III) калия массовой концентрации 10 г/дм3;
10,06 и 0,0175 - поправочные коэффициенты: установленные эмпирическим путем для 10 см3 раствора гексацианоферрата (III) калия массовой концентрации точно 10 г/дм3;
У - объем испытуемого раствора, израсходованный при окончательном титровании, см3;
А - фактор разведения навески
![]()
a - массовая доля углеводов в луке, %;
0,95 - коэффициент пересчета инвертного сахара на сахарозу.
Вычисление производят до второго десятичного знака, результат округляют до первого десятичного знака.
За конечный результат испытания принимают среднее арифметическое результатов двух параллельных определений, допустимое расхождение между которым не должно превышать 0,5 %, а между результатами определений в 2-х разных лабораториях - не более 1 %.
4.7.3.2. Определение углеводов в луке репчатом цианидным методом
Метод предназначен для определения массовой доли углеводов сырого репчатого лука, используемого для приготовления исследуемых фаршей изделий.
Аппаратура, материалы, реактивы. Указаны на с. 74, 76.
Подготовка к испытанию. Среднюю пробу лука репчатого (3 - 4 шт.) очищают, срезая у луковицы донце и шейку, удаляют сухие листья, моют, обсушивают, измельчают на мясорубке с мелкой решеткой и тщательно растирают в ступке до однородной массы.
Проведение испытания. Навеску подготовленной пробы продукта массой 15 г берут в стакан с точностью до 0,01 г, предварительно перемешав пробу во избежание отделения сока. Навеску переносят в мерную колбу вместимостью 200 см3 с помощью 100 - 200 см3 дистиллированной воды, нагретой до 40 - 50 °C. Органические кислоты, содержащиеся в луке, нейтрализуют раствором карбоната натрия с массовой долей 15 % до посинения красной лакмусовой бумаги. Затем колбу нагревают на водяной бане с температурой 80 °C в течение 30 мин, охлаждают до температуры 20 °C и осаждают несахара, добавляя по 10 см3 раствора гексацианоферрата (II) калия массовой концентрации 150 г/дм3 и раствора сульфата цинка массовой концентрации 300 г/дм3, хорошо перемешивают и доливают водой до метки. Если жидкость над осадком недостаточно прозрачна, количество осадителей увеличивают в 1,5 - 2 раза. Осадок отстаивают 50 - 60 мин, затем раствор фильтруют в сухую колбу. Фильтрат должен быть прозрачным.
Инверсию сахарозы проводят, как указано на с. 218. В полученном растворе определяют массовую долю углеводов цианидным методом (см. с. 56).
Обработка результатов анализа. Массовую долю углеводов (a, %) в луке репчатом рассчитывают по формуле:
|
|
(97) |
где V1 - объем испытуемого раствора, израсходованный при окончательном титровании, см3.
Остальные обозначения те же, что в формуле (96).
Вычисления проводят до второго десятичного знака, результаты округляют до первого десятичного знака.
За конечный результат принимают среднее арифметическое значение двух параллельных определений, допустимое расхождение между которыми не должно превышать в одной лаборатории 0,5 %, в 2-х разных лабораториях - 1,0 %.
Раздел 5. Контроль качества булочных и мучных кондитерских изделий
5.1. Сдобные булочные изделия
5.1.1. Отбор проб и подготовка их к испытанию
Отбор проб для физико-химических испытаний описан в прил. 1, 2. При подготовке проб отобранные изделия освобождают от включений (повидла, изюма, орехов, кроме мака) и измельчают ножом или в размельчителе тканей. Если стандартами или ТУ нормируется влажность, массовая доля сахара, жира в мякише, то изделия освобождаются от корок (за исключением слойки) толщиной около 1 см.
Изделия, покрытые помадой (пирог с маком, баба ромовая и т.д.). Если по НТД влажность нормируется с учетом промочки, помады, то изделие измельчается целиком с предварительным удалением включений (кроме мака).
Массовую долю сахара и жира в выпеченных полуфабрикатах определяют после удаления корочки (в мякише).
Физико-химические показатели определяют не ранее 3 ч и не позднее 16 ч после выпечки.
Влажность определяют в соответствии с ГОСТ 21094-76 (см. с. 13).
Кислотность определяют в соответствии с ГОСТ 5670-51 (см. с. 78).
Массовую долю жира определяют в соответствии с ГОСТ 5668-68, рефрактометрическим (см. с. 25) и арбитражным (см. с. 29) методами.
Массовую долю сахара определяют в соответствии с ГОСТ 5672-68 после гидролиза сахарозы и выражают его в сахарозе на сухое вещество. Массу навески изделия рассчитывают по формуле (74). Приготовление раствора редуцирующих сахаров из навески до и после инверсии сахарозы проводят, как указано на с. 148. Редуцирующие сахара определяют перманганатным методом (см. с. 47).
5.2. Мучные кондитерские изделия
5.2.1. Отбор проб и подготовка их к испытанию
Отбор проб для контроля физико-химических показателей мучных кондитерских изделий проводят по ГОСТ 5904-82 (см. прил. 2). Масса пробы для лабораторных испытаний должна быть не менее 100 г.
При подготовке проб из весового кекса, бисквита, рулетов, коврижки из разных мест лабораторного образца вырезают небольшие порции и объединяют их. У кекса удаляют включения.
Штучные кексы массой до 400 г используют для анализа целиком, предварительно удалив включения.
При определении массовой доли сахара, жира и кислотности у кекса перед удалением включений дополнительно обрезают корки.
При подготовке ромовой бабы к определению влажности также удаляют включения.
Печенье, пряники, коржики отбирают из разных мест средней пробы.
Пробы для анализа измельчают в фарфоровой ступке, на терке или механическом измельчителе и немедленно помещают в закрываемую посуду. Перед взятием навесок пробу перемешивают.
Анализ выпеченных полуфабрикатов, кексов, коврижек, бисквита, пряников и т.п. проводят не ранее чем через 16 ч после изготовления.
В мучных кондитерских изделиях определяют массовые доли влаги (см. с. 12), сахара (см. с. 149), жира (см. с. 29), кислотность (см. с. 78), щелочность (см. с. 81).
Физико-химические показатели тортов и пирожных определяют в полуфабрикатах и в готовых изделиях без отделки кремом после выпечки (см. с. 149).
Раздел 6. Контроль качества продовольственного сырья
Основные физико-химические показатели, по которым исследуется качество продовольственного сырья, используемого для приготовления блюд и кулинарных изделий, а также ГОСТы на методы испытаний приведены в прил. 3.
Для оценки гигиенических показателей безопасности продовольственного сырья и пищевых продуктов используются унифицированные методы анализа, предусмотренные в общесоюзных санитарно-гигиенических и санитарно-противоэпидемических правилах и нормах, методических указаниях и рекомендациях Минздрава СССР, перечень которых приведен в прил. 4.
6.1. Определение качества меда натурального
Мед натуральный (ГОСТ 19792-87) по физико-химическим показателям должен соответствовать требованиям, указанным в табл. 48.
Таблица 48
Физико-химические показатели меда натурального
|
Наименование показателя |
Значение для меда |
||
|
всех видов, кроме меда с белой акации и хлопчатника |
с белой акации |
с хлопчатника |
|
|
Массовая доля воды, %, не более |
21 |
21 |
19 |
|
Массовая доля редуцирующих сахаров (к безводному веществу), %, не менее |
82 |
76 |
86 |
|
Массовая доля сахарозы (к безводному веществу), %, не более |
6 |
10 |
5 |
|
Диастазное число (к безводному веществу), ед. Готе, не менее |
7 |
5 |
7 |
|
Оксиметилфурфурол в 1 кг меда, мг, не более |
25 |
25 |
25 |
6.1.1. Определение массовой доли воды
Метод основан на зависимости показателя преломления меда от содержания в нем воды.
Аппаратура, материалы. Рефрактометр с ценой деления шкалы показателя преломления не более 1∙10-3; баня водяная; термометр лабораторный с диапазоном измерения 0 - 100 °C, ценой деления 1 °C; пробирки стеклянные диаметром 7 мм, высотой 30 - 40 мм.
Подготовка к испытанию. Для проведения испытания используют жидкий мед. Если мед закристаллизован, помещают около 1 см3 меда в пробирку, плотно закрывают резиновой пробкой и нагревают на водяной бане при температуре 60 °C до полного растворения кристаллов. Затем пробирку охлаждают до температуры воздуха в лаборатории. Воду, сконденсировавшуюся на внутренней поверхности стенок пробирки, и массу меда тщательно перемешивают стеклянной палочкой.
Проведение испытания. Одну каплю меда наносят на призму рефрактометра и измеряют показатель преломления при температуре 20 °C.
Замеры производят не менее двух раз, каждый раз нанося на призму испытуемый мед, и за конечный результат принимают среднее арифметическое значение двух параллельных определений.
Обработка результатов. Полученный показатель преломления меда пересчитывают на массовую долю воды в меде по табл. 49.
Таблица 49
|
Коэффициент рефракции |
Массовая доля воды, % |
Коэффициент рефракции |
Массовая доля воды, % |
Коэффициент рефракции |
Массовая доля воды, % |
|
1,5044 |
13,0 |
1,4935 |
17,2 |
1,4830 |
21,4 |
|
1,5038 |
13,2 |
1,4930 |
17,4 |
1,4825 |
21,6 |
|
1,5033 |
13,4 |
1,4925 |
17,6 |
1,4820 |
21,8 |
|
1,5028 |
13,6 |
1,4920 |
17,8 |
1,4815 |
22,0 |
|
1,5023 |
13,8 |
1,4915 |
18,0 |
1,4810 |
22,2 |
|
1,5018 |
14,0 |
1,4910 |
18,2 |
1,4805 |
22,4 |
|
1,5012 |
14,2 |
1,4905 |
18,4 |
1,4800 |
22,6 |
|
1,5007 |
14,4 |
1,4900 |
18,6 |
1,4795 |
22,8 |
|
1,5002 |
14,6 |
1,4895 |
18,8 |
1,4790 |
23,0 |
|
1,4997 |
14,8 |
1,4890 |
19,0 |
1,4785 |
23,2 |
|
1,4992 |
15,0 |
1,4785 |
19,2 |
1,4780 |
23,4 |
|
1,4987 |
15,2 |
1,4880 |
19,4 |
1,4775 |
23,6 |
|
1,4982 |
15,4 |
1,4875 |
19,6 |
1,4770 |
23,8 |
|
1,4976 |
15,6 |
1,4870 |
19,8 |
1,4765 |
24,0 |
|
1,4971 |
15,8 |
1,4865 |
20,0 |
1,4760 |
24,2 |
|
1,4966 |
16,0 |
1,4860 |
20,2 |
1,4755 |
24,4 |
|
1,4961 |
16,2 |
1,4855 |
20,4 |
1,4750 |
24,6 |
|
1,4956 |
16,4 |
1,4850 |
20,6 |
1,4745 |
24,8 |
|
1,4950 |
16,6 |
1,4845 |
20,8 |
1,4740 |
25,0 |
|
1,4946 |
16,8 |
1,4840 |
21,0 |
|
|
|
1,4940 |
17,0 |
1,4835 |
21,2 |
|
|
Если определения проводят при температуре ниже или выше 20 °C, то вводят поправку на каждый градус Цельсия: для температур выше 20 °C - прибавляют к показателю преломления 0,00023; для температур ниже 20 °C - вычитают из показателя преломления 0,00023.
Допустимые расхождения между результатами определений не должны превышать 0,1 %.
6.1.2. Определение массовой доли редуцирующих сахаров и сахарозы
Определение массовой доли сахаров в меде проводят по ГОСТ 19792-87.
6.1.3. Определение диастазного числа
Диастазное число характеризует активность амилолитических ферментов меда.
Метод основан на колориметрическом определении количества субстрата, расщепленного в условиях проведения ферментативной реакции, и последующем вычислении диастазного числа.
Диастазное число выражают количеством кубических сантиметров раствора крахмала массовой долей 1 %, которое разлагается за 1 ч амилолитическими ферментами, содержащимися в 1 г безводного вещества меда. 1 см3 раствора крахмала соответствует 1 единице активности.
Аппаратура, материалы, реактивы. Фотоэлектроколориметр; pH-метр; электрод измерительный стеклянный; баня-термостат водяная на 20 и 40 °C; пробирки стеклянные диаметром 20 мм и высотой 200 мм; бюретка вместимостью 25 см3 с ценой деления 0,1 см3; пипетки вместимостью 1, 2 и 5 см3; колбы мерные вместимостью 50 см3; секундомер; крахмал растворимый для йодометрии, раствор массовой долей 0,25 %; кислота уксусная ледяная плотностью 1,07 г/см3, раствор концентрации 0,2 моль/дм3; ацетат натрия трехводный, раствор концентрации 0,2 моль/дм3; хлорид натрия, раствор концентрации 0,1 моль/дм3; 2,4-динитрофенол; йод, раствор концентрации 0,25 моль/дм3; раствор буферный стандартный с pH, близким к 5,0, для проверки стеклянного электрода; вода дистиллированная.
Подготовка к испытанию. Ацетатный буферный раствор концентрации 0,2 моль/дм3 с pH 5,0 приготовляют, смешивая одну объемную часть раствора уксусной кислоты и три объемных части раствора ацетата натрия. В полученном буферном растворе растворяют 2,4-динитрофенол с таким расчетом, чтобы его концентрация в комбинированном реактиве составила 0,05 %. Проверяют pH раствора потенциометрически и в случае отклонений от pH 5,0 его корректируют, добавляя раствор уксусной кислоты 0,3 моль/дм3 или раствор ацетата натрия концентрации 0,2 моль/дм3.
Комбинированный реактив готовят из восьми объемных частей раствора крахмала, пяти объемных частей буферного раствора с 2,4-динитрофенолом и одной объемной части раствора хлорида натрия. При приготовлении комбинированного реактива в количестве, равном или большем 1 дм3, объем соответствующих растворов отмеривают с точностью до 0,5 см3. Полученную смесь тщательно встряхивают. Реактивы хранят при комнатной температуре не более 3 мес.
Раствор меда. 5 г меда, взвешенного с точностью до 0,01 г, растворяют в дистиллированной воде в мерной колбе вместимостью 50 см3. 1 см3 такого раствора содержит 0,1 г меда.
Раствор крахмала. 0,25 г крахмала, взвешенного с точностью до 0,001 г, размешивают в стаканчике вместимостью 50 см3 с 10 - 20 см3 дистиллированной воды и количественно переносят в коническую колбу, где не сильно кипит 80 - 90 см3 дистиллированной воды. Кипение продолжают 2 - 3 мин. Колбу охлаждают до 20 °C, содержимое количественно переносят в мерную колбу вместимостью 100 см3 и доводят до метки.
Проведение испытания. В сухую пробирку отмеривают из бюретки 14 см3 комбинированного реактива. Пробирку закрывают резиновой пробкой и помещают на 10 мин в водяную баню с температурой 40 °C. Затем в пробирку вносят пипеткой 1 см3 раствора меда. Содержимое перемешивают пятикратным перевертыванием, и пробирку вновь помещают на водяную баню, одновременно включая секундомер. Пробирку выдерживают на водяной бане в течение 15 мин. при температуре (40 ± 0,02) °С.
Пипеткой отбирают 2 см3 реакционной смеси, которую вносят при перемешивании в мерную колбу вместимостью 50 см3, содержащую 40 см3 воды и 1 см3 раствора йода, имеющих температуру 20 °C. Раствор доводят водой до метки. Колбу закрывают пробкой, содержимое хорошо перемешивают и выдерживают на водяной бане при 20 °C в течение 10 мин.
Одновременно проводят контрольный опыт, заменяя раствор меда дистиллированной водой.
Оптическую плотность измеряют на фотоэлектроколориметре против воды при светофильтре с длиной волны 582 или 590 нм, используя кювету с рабочей длиной 10 мм. Значения оптической плотности испытуемого раствора (Дисп.) и контрольного опыта (Дк) записывают с точностью 0,001.
Обработка результатов. Диастазное число меда (X, ед. Готе) в пересчете на 1 г безводного вещества вычисляют по формуле:
|
|
(98) |
где:
Дк - оптическая плотность контрольного раствора;
Дисп. - оптическая плотность испытуемого раствора;
80 - коэффициент пересчета;
W - массовая доля воды в меде, %.
За окончательный результат испытания принимают среднее арифметическое результатов двух параллельных определений. Допустимые расхождения между результатами параллельных определений не должны превышать 0,5 ед. Готе в интервале величин менее 10 ед.
6.1.4. Качественная реакция на оксиметилфурфурол
Метод основан на образовании в кислой среде соединения оксиметилфурфурола с резорцином, окрашенного в вишнево-красный цвет.
Материалы и реактивы. Ступки фарфоровые диаметром 70 мм с пестиком; чашки фарфоровые диаметром 50 мм; эфир этиловый; хлорид кальция 2-водный; резорцин; кислота соляная концентрированная плотностью 1,19 г/см3; натрий металлический плавленый.
Подготовка к испытанию. Безводный эфир: эфир выдерживают не менее двух суток с хлоридом кальция (200 г хлорида кальция на 1 дм3 эфира) и быстро фильтруют через бумажный фильтр в склянку оранжевого стекла; в склянку с эфиром помещают нарезанный кусочками металлический натрий (около 20 г на 1 дм3 эфира); эфир выдерживают с натрием до тех пор, пока внесение дополнительного кусочка натрия не будет сопровождаться выделением пузырьков газа; высушенный эфир хранят в склянке с притертой пробкой в прохладном и затемненном месте.
Раствор резорцина с массовой долей 1 %: 1 г резорцина растворяют в 100 см3 концентрированной соляной кислоты (раствор должен быть бесцветным) и хранят в прохладном месте в склянке оранжевого стекла с притертой пробкой.
Проведение испытания. В сухой фарфоровой ступке тщательно перемешивают пестиком в течение 2 - 3 мин около 3 г меда и 15 см3 эфира. Эфирную вытяжку переносят в сухую фарфоровую чашку и повторяют перемешивание меда с новой порцией эфира. Эфирные вытяжки объединяют и дают эфиру испариться под тягой при температуре не выше 30 °C. К остатку прибавляют 2 - 3 капли раствора резорцина.
Появление розового или оранжевого окрашивания в течение 5 мин свидетельствует о наличии оксиметилфурфурола.
6.1.5. Количественное определение оксиметилфурфурола
Метод основан на колориметрическом определении оксиметилфурфурола в присутствии барбитуровой кислоты и паратолуидина.
Аппаратура, материалы, реактивы. Весы лабораторные; фотоэлектроколориметр; баня водяная; часы песочные на 1 мин; электроплитка или газовая горелка; термометр лабораторный до 100 °C; колбы мерные вместимостью 50 см3; пробирки стеклянные с притертой пробкой, вместимостью 10 см3; пипетки вместимостью 1, 2, 5, 10 см3; барбитуровая кислота пара-толуидин; изопропанол; кислота уксусная ледяная плотностью 1,07 г/см3; вода дистиллированная; гексацианоферрат калия; сульфат цинка кристаллогидрат семиводный.
Подготовка к испытанию. Раствор барбитуровой кислоты: 500 мг барбитуровой кислоты, высушенной при 105 °C в течение 1 ч, переносят в колбу на 100 см3, добавляют 70 см3 дистиллированной воды, растворяют при нагревании в водяной бане, охлаждают до 20 °C и доводят до метки. Раствор можно хранить при охлаждении длительное время. В случае образования кристаллов раствор нагревают на водяной бане примерно до 60 °C до полного растворения кристаллов. Колба должна быть закрыта легко вынимаемой пробкой.
Раствор пара-толуидина: 10 г пара-толуидина при температуре 44 - 45 °C растворяют в 50 см3 изопропанола при слабом нагревании на водяной бане, переносят в мерную колбу вместимостью 100 см3, добавляют 10 см3 ледяной уксусной кислоты при перемешивании и при 20 °C доводят изопропанолом до метки. Раствор используют через 24 ч после приготовления, хранят в прохладном и темном месте не более 1 мес. Реактив Керреса: 15 г гексацианоферрата калия растворяют в дистиллированной воде в колбе вместимостью 100 см3; 20,4 г сульфата цинка кристаллогидрата растворяют в дистиллированной воде в колбе вместимостью 100 см3.
Раствор меда: (10,00 ± 0,01) г меда растворяют приблизительно в 20 см3 свежепрокипяченной и остывшей дистиллированной воды, количественно переносят в мерную колбу вместимостью 50 см3. Мутные растворы осветляют реактивом Керреса. Для этого в колбу добавляют одну каплю гексацианоферрата калия, перемешивают, добавляют одну каплю сульфата цинка и при 20 °C доводят водой до метки. Перемешивают и отфильтровывают через неплотный фильтр. Раствор используют немедленно.
Проведение испытания. В две чистые сухие пробирки наливают по 2 см3 раствора меда и 5 см3 пара-толуидина. В одну пробирку добавляют 1 см3 дистиллированной воды (контроль), перемешивают и содержимым этой пробирки заполняют кювету с толщиной слоя раствора 10 мм. Не позднее чем через 1 - 2 мин, во вторую пробирку приливают 1 см3 барбитуровой кислоты, перемешивают и заполняют вторую кювету.
Измерение производят на фотоэлектроколориметре со светофильтром с максимумом пропускания (540 ± 10) нм по отношению к контрольному раствору ежеминутно в течение 6 мин.
Обработка результатов. Оксиметилфурфурол (X, мг) в 1 кг меда вычисляют по формуле:
|
|
(99) |
где:
K - максимальное значение измеренной экстинкции;
S - толщина слоя жидкости в кювете колориметра, см;
19,2 - постоянный коэффициент экстинкции;
10 - коэффициент пересчета граммов меда в килограммы.
За окончательный результат испытания принимают среднее арифметическое результатов двух параллельных определений.
6.1.6. Экспресс-метод определения натуральности меда*(2)
________________
*(2) А. Авшалумова (Дагестанский сельскохозяйственный институт).
Один из методов определения натуральности меда - реакция на фосфор, применяемая для определения его содержания в растениях.
Аппаратура, материалы, реактивы. Весы лабораторные; колбы мерные вместимостью 100 см3; цилиндры мерные на 10; 50; 100 см3; фильтры; молибденовокислый аммоний, раствор массовой долей 6 %; азотная кислота концентрированная плотностью 1,4 г/см3; раствор бензидина; кислота уксусная ледяная плотностью 1,07 г/см3; ацетат натрия; дистиллированная вода.
Подготовка к испытанию. Раствор молибденовокислого аммония: к 100 см3 молибденовокислого аммония, раствор массовой долей 6 %, добавляют 35 см3 концентрированной азотной кислоты.
Раствор бензидина: 0,05 г бензидина растворяют в 10 см3 ледяной уксусной кислоты, переносят в мерную колбу емкостью 100 см3 и доливают дистиллированной водой до метки.
Насыщенный раствор ацетата натрия: 31 - 32 г ацетата натрия растворяют в 100 см3 дистиллированной воды.
Все реактивы необходимо хранить в темной посуде.
Проведение испытания. На фильтровальную бумагу наносят каплю раствора молибденовокислого аммония с азотной кислотой (бумагу можно обработать заранее, подсушить на воздухе и хранить в темной посуде), затем на это же пятно наносят каплю раствора меда (1:2) и каплю раствора бензидина; вновь подсушивают бумагу и наносят каплю насыщенного раствора ацетата натрия. Если мед натуральный, то бумага приобретает синюю окраску, если искусственный, то этого не происходит.
Появление бледно-голубой окраски указывает на возможную фальсификацию. Интенсивность синей окраски можно определить по контрольной пробе из натурального меда на этом же листе бумаги.
6.1.7. Качественные реакции на обнаружение наполнителей в меде
Аппаратура, материалы, реактивы. Весы лабораторные; водяная баня; электрическая плитка; цилиндры вместимостью на 5, 25, 10 см3; пипетки вместимостью 1 см3; воронки; колбы конические вместимостью 250 см3; фильтры; раствор йода в йодиде калия; ацетат свинца, х.ч.; раствор танина с массовой долей 5 %; соляная кислота конц.
Обнаружение муки, крахмала и других порошкообразных веществ.
Мед растворяют в трех-, пятикратном объеме воды. При наличии примесей они оседают на дно. При добавлении в раствор меда 1 - 2 капель раствор йода в иодиде калия он посинеет при наличии в нем муки или крахмала.
Обнаружение свекольной патоки. К 5 см3 раствора меда с массовой долей 20 % прибавляют 2,5 г ацетата свинца и 22,5 см3 метилового спирта. Образование желтоватого осадка указывает на фальсификацию меда патокой.
Обнаружение крахмальной патоки. Метод основан на осаждении спиртом крахмальных декстринов. Растворяют 5 г меда в 10 см3 воды, нагревают на водяной бане и добавляют 0,5 см3 раствора таинина с массовой долей 5 %, затем взбалтывают и фильтруют.
Для удержания в растворе медовых декстринов в раствор добавляют по 2 капли соляной кислоты (плотностью 1,19 г/см3) на каждый кубический сантиметр исследуемого медового раствора.
Помутнение жидкости от прибавления десятикратного количества этилового спирта указывает на присутствие крахмальной патоки.
Раздел 7. Контроль правильности проведения технологического процесса
7.1. Определение эффективности тепловой обработки мясных и рыбных кулинарных изделий
7.1.1. Проба на пероксидазу
Метод основан на способности фермента пероксидазы принимать участие в процессах окисления за счет кислорода пероксида водорода. Присутствие пероксидазы устанавливают, используя реакции с гваяколом, бензидином, амидопирином (пирамидоном). При температуре 80 °C пероксидаза инактивируется. Следовательно, если в исследуемом изделии пероксидаза обнаруживается, тепловая обработка считается недостаточной.
Аппаратура, материалы, реактивы. Весы лабораторные; пробирки химические диаметром 15 мм; пробки корковые; штатив для пробирок; ступка фарфоровая диаметром 7 - 9 см; капельницы; часы песочные на 1, 2 мин; воронки стеклянные диаметром 4 - 5 см; пипетки вместимостью 1 и 20 см3 колбы конические вместимостью 50 и 100 см3; бумага фильтровальная; вата гваякол, спиртовой раствор с массовой долей 1 % (1 г гваякола растворяют этиловым спиртом в мерной колбе на 100 см3); бензидин, спиртовой раствор с массовой долей 0,02 % (20 мг бензидина растворяют в 100 см3 этилового спирта); амидопирин, спиртовой раствор с массовой долей 2 % (2 г амидопирина растворяют в 98 см3 этилового спирта); спирт этиловый; пероксид водорода (30 - 35 %), раствор с массовой долей 10 %; кислота уксусная ледяная; ацетат натрия безводный; вода дистиллированная.
Проведение испытания. Окислительно-восстановительные свойства пероксидазы проявляются в строго определенном интервале pH. Наиболее интенсивная окраска наблюдается в интервале значений pH от 4,4 до 6,9; менее интенсивная при pH 3,4 и выше; не проявляется при pH выше 10,4.
При анализе используют ацетатный буферный раствор с pH 4,9.
Измельченную навеску, взятую из внутренней части жареного изделия в количестве 10 г и взвешенную с точностью до 0,01 г, растирают в ступке с 20 см3 дистиллированной воды и фильтруют через бумажный фильтр или слой ваты в коническую колбу. Затем отбирают в пробирку 0,5 см3 фильтрата, добавляют 0,5 см3 ацетатного буфера, 0,5 см3 спиртового раствора гваякола, 0,25 см3 свежеприготовленного раствора пероксида водорода и встряхивают. При достаточной термической обработке мясного изделия раствор остается бесцветным, при недостаточной, в зависимости от количества сохраненной пероксидазы, окраска может быть от светло-голубой до темно-синей и проявляется в течение 1 мин.
При использовании спиртового раствора бензидина или спиртового раствора амидопирина в пробирку отбирают 1 см3 фильтрата, добавляют 1 см3 одного из указанных растворов, а также 0,5 см3 раствора пероксида водорода и встряхивают. При наличии пероксидазы в течение 1 мин появляется соответственно сине-зеленое или сине-фиолетовое окрашивание. При достаточной тепловой обработке изменения цвета не происходит.
Учитывая, что в мясе больных животных и в несвежем мясе происходит инактивация фермента пероксидазы, для окончательного суждения о качестве тепловой обработки кулинарных изделий необходимо проверить наличие пероксидазы в мясном полуфабрикате. При отсутствии пероксидазы в полуфабрикате достаточность тепловой обработки определяют пробой на фосфатазу.
7.1.2. Проба на фосфатазу
Качественная реакция. Метод основан на способности фермента фосфатазы расщеплять бариевую соль паранитрофенилфосфата при температуре 38 °C, освобождая паранитрофенол, который окрашивает среду в желтый цвет.
Аппаратура, материалы, реактивы. Весы лабораторные; плитка электрическая; баня водяная; ступка фарфоровая диаметром 7 - 9 см; цилиндр вместимостью 1 см3; воронка делительная вместимостью 250 см3; пробки корковые; капельница; воронки стеклянные диаметром 4 - 5 см; марля; бумага фильтровальная; вата стеклянная; бариевая соль паранитрофосфата, насыщенный раствор; гидроксид натрия, раствор массовой концентрации 400 г/дм3 (Д = 1,43 г/см3); хлорид магния, раствор массовой концентрации 5 г/дм3; ацетатный буфер pH 5,4; вода дистиллированная.
Проведение испытания. Измельченную навеску, взятую из внутренней части изделия в количестве 20 г и взвешенную с точностью до 0,01 г, переносят в ступку и растирают, добавляя постепенно 50 см3 дистиллированной воды. Полученную взвесь процеживают через двойной слой марли, а оставшуюся в марле навеску отжимают, затем вытяжку фильтруют через сухой складчатый фильтр и делят пополам. Одну часть (фильтрат 1) исследуют непосредственно, другую (фильтрат 2) переносят в коническую колбу, доводят до кипения и снова фильтруют - эта часть фильтрата является контрольной.
Для проверки активности фосфатазы в пробирку отмеривают 1 см3 фильтрата 1, прибавляют 2 капли раствора хлорида магния массовой концентрации 5 г/дм3, 2 капли ацетатного буфера (pH 5,4) и 0,5 см3 раствора бариевой соли паранитрофенилфосфата.
Для контроля во вторую пробирку отмеривают 1 см3 фильтрата 2 и добавляют те же реактивы, что и в первую. Обе пробирки помещают на 1 ч в водяную баню или термостат при температуре 37 - 38 °C. Затем в обе пробирки добавляют по капле раствора гидроксида натра.
При достаточной тепловой обработке кулинарного изделия окраска в обеих пробирках не меняется. При недостаточной тепловой обработке раствор желтеет.
Определение остаточной активности кислой фосфатазы (количественное определение). Метод основан на фотометрическом определении в продукте интенсивности развивающейся окраски, зависящей от остаточной активности кислой фосфатазы, выраженной массовой долей фенола.
Аппаратура, материалы, реактивы. Весы лабораторные; потенциометр с погрешностью измерения ±0,06 pH; фотоэлектроколориметр или спектрофотометр для измерения в видимой области спектра; ультратермостат или водяная баня; воронки; колбы мерные вместимостью 500 и 1000 см3; пипетки градуированные на 1; 5; 10 см3; палочки стеклянные; пробирки; бумага фильтровальная; груша резиновая; кислота лимонная; цитрат натрия 5-водный; динатриевая соль фенилфосфорной кислоты, раствор массовой концентрации 2 г/дм3, свежеприготовленный; кислота трихлоруксусная, кристаллическая, растворы массовой концентрации 50 и 200 г/дм3; гидроксид натрия, раствор C(NaOH) = 0,5 моль/дм3; вода дистиллированная; фенол; толуол; вольфрамат натрия; сульфат лития 1-водный; кислота ортофосфорная плотностью 1,72 г/см3; кислота соляная плотностью 1,19 г/см3; бром.
Подготовка к испытанию. Ацетатный буфер: в мерной колбе вместимостью 1000 см3 в дистиллированной воде растворяют 13,88 г цитрата натрия и 0,588 г лимонной кислоты, доливают водой до метки и перемешивают, pH буфера 6,5. Затем добавляют 1 см3 толуола. Раствор хранят в холодильнике при температуре 4 ± 1 °C не более 12 сут.
Реактив Фолина: 100 г вольфрамата натрия и 25 г молибдата натрия растворяют в 700 см3 дистиллированной воды. К раствору добавляют 50 см3 ортофосфорной кислоты и 100 см3 соляной кислоты. Смесь осторожно кипятят в течение 10 ч в колбе вместимостью 2000 см3 с обратным холодильником, после чего охлаждают и добавляют 150 г сульфата лития, 50 см3 воды и несколько капель брома. Остаток брома отгоняют кипячением смеси без холодильника в вытяжном шкафу, охлаждают, переносят в мерную колбу вместимостью 1000 см3, доводят объем дистиллированной водой до метки, перемешивают и фильтруют. Реактив должен быть золотисто-желтого цвета без зеленого оттенка; его хранят в склянке с притертой пробкой в темном месте не более 6 мес.
Стандартный раствор: 2 г фенола (взвешивают с точностью до 0,001 г) растворяют в воде в мерной колбе вместимостью 1000 см3, доводят объем до метки и перемешивают. Отбирают пипеткой с помощью резиновой груши 5 см3 раствора в колбу вместимостью 500 см3, добавляют около 300 см3 дистиллированной воды, вносят 25 г кристаллической трихлоруксусной кислоты. После растворения содержимое колбы доводят до метки дистиллированной водой и перемешивают. Полученный раствор содержит 20 мкг фенола в 1 см3.
Построение градуировочного графика. В пробирки вносят следующие объемы стандартного раствора: 0; 0,25; 0,5; 1,0; 1,5; 2,0 см3, что соответствует массе фенола: 0; 5; 10; 20; 30; 40 мкг. Доводят объем каждой пробирки до 2,5 см3, добавляя соответствующий объем раствора трихлоруксусной кислоты массовой концентрации 50 г/дм3 (2,5; 2,25; 2,0; 1,5; 1,0; 0,5 см3), и перемешивают. В каждую пробирку добавляют 5 см3 раствора гидроксида натрия, перемешивают, выдерживают 10 мин, добавляют 1,5 см3 реактива Фолина, разведенного дистиллированной водой в соотношении 1:2, и перемешивают.
Через 30 мин измеряют оптическую плотность растворов по отношению к раствору трихлоруксусной кислоты массовой концентрации 50 г/дм3 на фотоэлектроколориметре с применением светофильтра с длиной волны 600 ± 10 нм в кювете с расстоянием между рабочими гранями 10 мм или спектрофотометра при длине волны 600 нм в кювете аналогичного размера.
По полученным средним данным по трем стандартным растворам на миллиметровой бумаге размером 20×20 см строят градуировочный график. На оси абсцисс откладывают значение массовой доли фенола (микрограмм в 9 см3 окрашенного раствора); на оси ординат - значение соответствующей оптической плотности (Д). Градуировочный график должен проходить через начало координат (рис. 5).
Рисунки должны быть выполнены сначала на миллиметровке и затем на кальке.
Рис. 5. Градуировочный график для определения массовой доли
фенола
с помощью фотоэлектроколориметра (пример)
Проведение испытания. От объединенной пробы, подготовленной к испытанию, берут 2 навески массой по 1 г (с точностью до 0,001 г) и переносят в две пробирки (контрольную и опытную).
В пробирки вносят по 10 см3 ацетатного буфера pH 6,5, тщательно перемешивают стеклянной палочкой и настаивают в течение 20 мин при температуре 20 °C, периодически перемешивая.
В контрольную пробирку добавляют 5 см3 200 г/дм3 раствора трихлоруксусной кислоты, перемешивают и добавляют 5 см3 2 г/дм3 раствора динатриевой соли фенилфосфорной кислоты, выдерживают 10 мин и фильтруют.
В опытную пробирку добавляют 5 см3 2 г/дм3 раствора динатриевой соли фенилфосфорной кислоты и помещают в ультратермостат при температуре 39 ± 1 °C на 1 ч, затем добавляют 5 см3 200 г/дм3 раствора трихлоруксусной кислоты, выдерживают 10 мин и фильтруют.
Для проведения цветной реакции из контрольной и опытной пробирок отбирают по 2,5 см3 безбелкового фильтрата. Цветную реакцию проводят по методу, описанному на с. 235.
Массу фенола в навеске определяют по градуировочному графику.
Обработка результатов. Массовую долю фенола (X, %) вычисляют по формуле:
|
|
(100) |
где:
m1 - масса фенола в опытной пробирке, найденная по градуировочному графику, мкг;
m2 - масса фенола в контрольной пробирке, найденная по градуировочному графику, мкг;
m - масса анализируемой пробы, г;
106 - коэффициент пересчета;
20 - разведение;
2,5 - объем фильтрата, отобранный для цветной реакции, см3.
Вычисление проводят до 0,0001.
За окончательный результат испытания принимают среднее арифметическое результатов двух параллельных определений, допустимое расхождение между которыми при P = 0,95 не должно превышать 10 % по отношению к среднему арифметическому.
Окончательный результат определяют до 0,001.
7.2. Контроль качества фритюрного жира
При продолжительной жарке продуктов во фритюре качество фритюрных жиров изменяется: жиры темнеют, приобретают резкий неприятный запах, горький привкус. В жире накапливаются вторичные термостабильные продукты окисления и сополимеризации, количество которых не должно превышать 1,0 %. Жир с массовой долей продуктов окисления более 1 % считается непригодным для пищевых целей. Лабораторный контроль качества фритюра осуществляется по органолептическим и физико-химическим показателям.
Для лабораторного контроля отбирают предварительно отфильтрованные пробы жиров (исходного и использованного для фритюрной жарки) в количестве 50 г каждого в посуду с притертыми пробками.
Качество фритюра определяют по органолептическим показателям ежедневно после окончания жарки.
Если жарка производится на неспециализированном оборудовании (электросковородах с непосредственным и косвенным обогревом, универсальных газовых жаровнях), то доброкачественность фритюра контролируется лабораторным путем через каждые 7 ч его использования.
Органолептическую оценку фритюрного жира проводят, пользуясь оценочной шкалой качества (табл. 50).
Если по органолептическим показателям фритюр получил оценку ниже трех баллов, лаборатория дает заключение о непригодности жира и по физико-химическим показателям его уже не оценивают.
Если при органолептической оценке жир получил оценку "удовлетворительно", то производят определение степени термического окисления физико-химическими методами.
7.2.1. Качественная проба на степень термического окисления фритюра из смесей жиров или подсолнечного масла
Цветная реакция основана на взаимодействии окисленных веществ, перешедших из фритюрного жира в спиртовой раствор гидроксида калия с метиленовым голубым. При содержании в исследуемом жире окисленных веществ до 1 % проба после добавления соответствующих реактивов приобретает розовый цвет, а свыше 1 % - желто-коричневый.
Аппаратура, материалы, реактивы. Пробирки химические из бесцветного стекла с внутренним диаметром 10 мм; колба коническая вместимостью 50 см3; капельница стеклянная лабораторная; штатив для пробирок; воронка стеклянная; пипетка вместимостью 1 см3; бумага фильтровальная крупнопористая; спиртовой раствор гидроксида калия с массовой долей 2 % (2 г едкого кали растворяют в этиловом спирте, помещают в колбу на 100 см3 и доводят до метки спиртом); спирт этиловый; метиленовый голубой, водный раствор с массовой долей 0,01 % (10 мг метиленового голубого растворяют в 100 см3 воды).
Проведение испытания. В пробирку с внутренним диаметром 10 мм помещают 3 см3 испытуемого подсолнечного масла или растопленного фритюрного жира, добавляют 7 см3 спиртового раствора гидроксида калия с массовой долей 2 %. Пробирку закрывают корковой (не резиновой) пробкой и энергично встряхивают 30 с. После разделения жидкостей верхний слой спиртово-щелочной вытяжки фильтруют через бумажный фильтр в колбочку. Для проведения реакции берут пипеткой 1 см3 фильтрата, помещают в пробирку и добавляют 5 капель метиленового голубого. Содержимое пробирки встряхивают и оставляют на 5 мин.
7.2.2. Определение степени термического окисления фритюрного жира по показателю преломления
Метод основан на сравнении показателя преломления фритюра и исходного свежего масла при температуре 20 °C и применим только для растительных масел, используемых для жарки пирожков (пончиков)*(3).
_______________
*(3) При использовании данного метода не определяют степень термического окисления фритюра (растительного масла) колориметрическим методом.
Установлено, что по мере накопления в масле продуктов окисления и сополимеризации возрастает показатель преломления жира. Разница между показателем преломления фритюра и исходного (свежего) масла не должна превышать 0,001.
Аппаратура, материалы, реактивы. Рефрактометр лабораторный; термостат; воронки стеклянные диаметром 3 - 4 см; стаканы химические вместимостью 25 - 50 см3; палочка стеклянная; марля; бумага фильтровальная; вода дистиллированная; спирт этиловый; эфир этиловый.
Проведение испытания. На центральную часть поверхности нижней призмы рефрактометра, предварительно установленного по дистиллированной воде, наносят 1 - 2 капли профильтрованного через крупнопористую фильтровальную бумагу исходного (свежего) масла. После замера показателя преломления призмы вытирают марлей, смоченной спирто-эфирной смесью (1:1), а затем сухой. На призму рефрактометра наносят 1 - 2 капли масла, использовавшегося для жарки пирожков (или пончиков). Определение показателя преломления повторяют 2 - 3 раза, нанося каждый раз новые капли на призму рефракторметра. За результат берут среднюю арифметическую величину.
Обработка результатов. Если показатель преломления определяют при температуре выше или ниже 20 °C, то вводят поправку на каждый градус отклонения температуры. Показатель преломления приводят к температуре 20 °C по следующей формуле:
|
|
(101) |
где:
![]() - искомый показатель преломления при
20 °C;
- искомый показатель преломления при
20 °C;
![]() ПД -
показатель преломления при температуре опыта;
ПД -
показатель преломления при температуре опыта;
t° - температура опыта;
0,00035 - изменение показателя преломления при изменении температуры на 1 °C.
Разность между показателями преломления фритюра и исходного свежего масла не должна превышать 0,0010.
Оценочная шкала качества фритюрных жиров
|
Жиры |
Количество баллов |
Показатели качества |
||
|
цвет в проходящем и отраженном свете на белом фоне |
вкус (при 40 °C) |
запах (при температуре не ниже 50 °C) |
||
|
коэффициент влажности |
||||
|
3 |
2 |
2 |
||
|
Фритюрный, Белорусский, Украинский, Восточный, салорастительное |
5 |
От белого до светло-желтого |
Для жира фритюрного и сала растительного - без постороннего привкуса; для жиров Белорусского, Украинского, Восточного - характерный для добавленного жира (соответственно говяжьего, свиного или бараньего) без постороннего привкуса |
Для жира фритюрного и сала растительного - без постороннего запаха; для жиров Белорусского, Украинского и Восточного - характерный для добавляемого жира без постороннего запаха |
|
То же |
4 |
Желтый |
Хороший, но с посторонним привкусом |
Со слабым посторонним запахом |
|
То же |
3 |
Желтый с коричневым оттенком |
Слабовыраженный, горьковатый |
Слабовыраженный, неприятный, продуктов термического распада жира |
|
То же |
2 |
Светлокоричневый |
Горький с ярко выраженным посторонним привкусом |
Ярко выраженный, неприятный, продуктов термического распада жира |
|
То же |
1 |
Коричневый |
Очень горький, вызывающий неприятное ощущение першения |
Резкий, неприятный, продуктов термического распада |
|
Подсолнечное масло |
5 |
Соломенно-желтый |
Без постороннего привкуса |
Без постороннего запаха |
|
То же |
4 |
Интенсивно-желтый |
Хороший, но с посторонним привкусом |
Без постороннего запаха |
|
То же |
3 |
Интенсивно-желтый с коричневым оттенком |
Слабовыраженный, горьковатый |
Слабовыраженный, неприятный, продуктов термического распада |
|
То же |
2 |
Светлокоричневый |
Горький с ярко выраженным посторонним привкусом |
Выраженный, неприятный, продуктов термического распада масла |
|
То же |
1 |
Коричневый или темнокоричневый |
Очень горький, вызывающий неприятное ощущение першения |
Резкий, неприятный, продуктов термического распада масла |
7.3. Расчет содержания сухих веществ и жира по рецептурам блюд и изделий
Основными показателями полноты вложения сырья в блюдо (изделие) являются содержание сухих веществ и жира.
Результаты анализов по этим показателям сравнивают с расчетными данными по рецептуре (теоретически максимальными) или с расчетными данными по рецептуре с учетом потерь сухих веществ и жира в процессе приготовления пищи, допустимых отклонений при порционировании и с учетом погрешности ускоренных или упрощенных методов исследования, а также техники ведения анализа (минимально допустимыми).
Максимальным (теоретическим) содержанием сухих веществ называют сумму сухих веществ сырьевого набора (по рецептуре) и введенной в блюдо поваренной соли (г).
Весь набор сырья по рецептуре выписывают массой нетто. Если в рецептуре набор сырья указан массой брутто, то его пересчитывают на массу нетто в соответствии с нормами отходов (приложения Сборника рецептур блюд и кулинарных изделий, 1981 г.). Затем для каждого из продуктов по таблицам справочника "Химический состав пищевых продуктов", 1987 г., находят процентное содержание сухих веществ и пересчитывают их на массу продуктов по рецептуре. Далее находят общую сумму сухих веществ в граммах.
При расчете супов, приготовленных на бульоне (мясо-костном или костном), к сухим веществам набора сырья прибавляют сухие вещества бульонов: для мясного - 5,1 и 3,6 г на порцию 500 г соответственно по II и III колонкам, для костного 5,7 и 3,7 г на порцию по II и III колонкам рец. № 174 Сборника рецептур блюд и кулинарных изделий, 1981 г.
Максимальное (теоретическое) содержание (Хмакс, г) сухих веществ в блюде (изделии) рассчитывают по формуле:
|
(102) |
где:
Со - количество сухих веществ в порции блюда (изделия), рассчитанное по рецептуре и таблицам химического состава пищевых продуктов, г;
С - содержание соли, г, обычно принимают: для первых блюд - 3 г (на 500 г), молочных супов - 2 г (на 500 г), вторых - 2 г (на 150 - 200 г), молочных каш - 1 г (на 150 - 200 г), салатов - 2 - 3 г (на 100 - 150 г), для соусов - 0,5 г (на 50 г).
Минимально допустимое содержание сухих веществ (Хмин, г) в порции блюда (изделия) рассчитывают по следующим формулам:
для первых блюд и соусов:
|
Хмин = 0,85(Со + С), |
(103) |
для холодных, вторых блюд, гарниров, сладких блюд и горячих напитков (кроме кофе и какао с молоком):
|
Хмин = 0,9(Со + С), |
(104) |
где:
0,85; 0,9 - коэффициенты, учитывающие потери сухих веществ в процессе приготовления и допустимые отклонения при порционировании блюд;
Со и С - обозначения, как в формуле (102).
При исследовании пудингов, сладких каш расчет фактического и максимального содержания сухих веществ ведут на массу без включений (изюма, цукатов, орехов).
Если найденное при анализе количество сухих веществ в блюде меньше минимально допустимого, значит, имеет место недовложение сырья. Превышение же максимально теоретического содержания сухих веществ будет указывать на то, что было вложено большее количество продуктов или допущено неправильное порционирование.
Для проверки правильности вложения жира по рецептуре и таблицам химического состава пищевых продуктов определяют суммарное количество чистого жира, введенного в блюдо с различными жировыми продуктами (маслом, сметаной и др.), т.е. находят максимально возможное содержание чистого жира в блюде. В процессе приготовления и порционирования блюд часть жира теряется, поэтому вводят поправку на потери жира: производственные и обусловленные погрешностью методов его определения.
Размеры потерь жира в зависимости от методов, использованных для его определения, приведены в табл. 8.
Вычитая потери из максимального количества жира в блюде, получают минимально допустимое содержание чистого жира, с которым сравнивают фактическое его содержание, полученное при анализе.
В супах, приготовленных на мясо-костном и костном бульонах, минимально допустимое содержание жира по рецептуре не рассчитывают, а фактическое содержание жира сравнивают с теоретическим.
Пример расчета 1. Анализировали суп картофельный с горохом, приготовленный по рец. 221 Сборника рецептур, 1981 г., приведенной в табл. 51.
Определено: масса блюда 480 г, масса блюда после упаривания
- 230 г. Масса навески для определения сухих веществ 5 г, масса высушенной
навески - 1,2 г. Количество сухих веществ в исследуемой порции 55,2 г ![]() в порции с выходом 500 г - 57,5 г
в порции с выходом 500 г - 57,5 г ![]() . Количество жира в 500 г супа 4,6 г.
Жир определяли экстракционно-весовым методом.
. Количество жира в 500 г супа 4,6 г.
Жир определяли экстракционно-весовым методом.
Количество сухих веществ, рассчитанное по таблицам химического состава пищевых продуктов, равно 84,99 г (табл. 51).
|
Продукты |
Масса нетто, г |
Количество сухих веществ, г |
|
|
в 100 г продукта, % |
в наборе сырья, г |
||
|
Картофель |
125 |
25 |
31,2 |
|
Морковь |
20 |
11,5 |
2,3 |
|
Лук репчатый |
20 |
14 |
2,8 |
|
Петрушка |
5 |
15 |
0,7 |
|
Горох |
50 |
86,0 |
43,0 |
|
Жир свиной топленый |
5 |
99,7 |
4,99 |
|
Итого: |
|
|
84,99 |
Свиной топленый жир содержит 99,7 г
чистого жира. Потери жира составляют 0,5 г ![]() . Минимально допустимое количество
сухих веществ в супе равно 74,8 г [0,85∙(84,99 + 3)], минимально
допустимое содержание чистого жира - 4,5 г.
. Минимально допустимое количество
сухих веществ в супе равно 74,8 г [0,85∙(84,99 + 3)], минимально
допустимое содержание чистого жира - 4,5 г.
Заключение. Масса порции супа ниже нормы на 20 г. Содержание сухих веществ ниже нормы на 17,3 г (74,8 - 57,5). Содержание жира в супе в норме.
Пример расчета 2. На анализ доставлены котлеты картофельные со сметаной, приготовленные по рец. № 357 (II колонка) Сборника рецептур блюд и кулинарных изделий, 1981 г., приведенной в табл. 52.
Таблица 52
|
Продукты |
Масса нетто, г |
Количество сухих веществ, г |
Количество жира, г |
||
|
в 100 г продукта, % |
в наборе сырья, г |
в 100 г продукта, % |
в наборе сырья, г |
||
|
Картофель |
215 |
25,0 |
53,75 |
- |
- |
|
Яйца |
6 |
26,0 |
1,56 |
11,5 |
0,69 |
|
Сухари пшеничные |
12 |
88,0 |
10,56 |
- |
- |
|
Жир кулинарный |
10 |
99,7 |
9,97 |
99,7 |
9,97 |
|
Масса жареных котлет |
200 |
- |
75,84 |
- |
10,66 |
|
Сметана |
20 |
27,3 |
5,46 |
20 |
4,00 |
|
Выход |
220 |
- |
81,30 |
- |
14,66 |
При анализе установлено: масса порции блюда 211 г, содержание сухих веществ 74,0 г, содержание жира 11,2 г.
Подсчитаем минимально допустимое содержание чистого жира в блюде, если анализ проводили методом Гербера.
Согласно табл. 8 жир в котлетах овощных определяется в количестве не менее 75 % от вложенного чистого жира по рецептуре, что составляет:
![]()
С учетом жира сметаны минимально допустимое содержание жира в блюде составит: 8,0 + 4,0 = 12 г, максимальное - 14,66 г.
Максимальное содержание сухих веществ в блюде (содержание поваренной соли 2 г):
Хмакс = 81,3 + 2 = 83,3 г.
Минимально допустимое содержание сухих веществ:
Хмин = 0,9∙(81,3 + 2) = 74,97, или 75,0 г.
Следовательно, Хмакс = 83,3 г; Хмин = 75,0 г.
Дополнительно отобрана для анализа сметана. Содержание жира в сметане, установленное анализом, - 20,0 %.
Фактическая средняя масса жареной котлеты, полученная взвешиванием 10 изделий, отобранных с противня, 98 г.
Заключение. Недовес порции составляет 213 - 211 = 2 г (норма 220 ± 7); содержание сухих веществ ниже нормы на 1,0 г (75,0 - 74,0), жира на 0,8 г (12,0 - 11,2). При массе двух котлет 196 г (в среднем) на порцию недовложение сухих веществ и жира объясняется недовложением сметаны при порционировании.
Пример расчета 3. На анализ доставлен клюквенный кисель, приготовленный по рец. № 934 Сборника рецептур блюд и кулинарных изделий, 1981 г., приведенной в табл. 53.
Таблица 53
|
Наименование продуктов |
Масса нетто, г |
Количество сухих веществ, г |
|
|
100 г продукта, % |
в наборе сырья, г |
||
|
Клюква |
20 |
10,5 |
2,10 |
|
Сахар |
20 |
99,86 |
19,97 |
|
Крахмал картофельный |
9 |
80,0 |
7,2 |
|
Выход |
200 |
- |
29,27, или 14,64 % |
Минимально допустимое содержание сухих веществ (X, г, %) в порции блюда (потери составляют 10 %):
X = 29,27∙0,9 = 26,34, или 13,17 %.
В результате анализа установлено: масса порции 210 г, среднее содержание сухих веществ по рефрактометру, определенное при температуре 23 °C, - 12,3 %. Поправка на температуру - 0,21 %. Содержание сухих веществ будет: 12,3 + 0,21 = 12,51 %.
Находим содержание сухих веществ (X, %) в блюде:
![]()
Заключение. Содержание сухих веществ в киселе ниже нормы на 0,7 % (13,17 - 12,51). Содержание сухих веществ в порции блюда в норме за счет порционирования (фактический выход порции 210 г).
7.4. Расчет рецептур полуфабрикатов и изделий по физико-химическим показателям
7.4.1. Расчет влажности теста, массовой доли сахара и жира в сдобных булочных, мучных кулинарных и кондитерских изделиях, мучных и отделочных полуфабрикатах для кондитерских изделий
Массовая доля жира и сахар нормируется ГОСТ, РСТ и ТУ на каждый вид изделия. Эти нормы являются гарантийными. При анализах допускаются отклонения от этих норм; они указаны в примечаниях к таблицам физико-химических показателей в каждой НТД. Если на какой-либо вид изделий нормативно-техническая документация отсутствует, то массовую долю жира и сахара определяют путем расчета по рецептуре. Для этого согласно таблицам химического состава пищевых продуктов (или НТД на сырье) считают сумму сухих веществ в граммах в сырье, входящем в рецептуру. Далее находят суммарное содержание чистого жира из компонентов сырья, содержание сахара и рассчитывают их процентное содержание в пересчете на сухое вещество. При этом принимают, что допускаемые отклонения содержания сахара и жира от расчетного по рецептуре в меньшую сторону не должны быть более указанных в табл. 54.
|
Наименование показателей |
Допускаемеые отклонения, % |
||||
|
до 5 |
5 - 10 |
10 - 20 |
20 - 30 |
30 и более |
|
|
Содержание сахара |
0,5 |
1,0 |
1 |
1,5 |
2 |
|
Содержание жира |
0,5 |
0,5 |
1 |
1,5 |
2 |
Пример расчета 1. На анализ доставлена ватрушка с творогом, приготовленная по рец. № 1098 Сборника рецептур блюд и кулинарных изделий, 1981 г.
Расход сырья для теста (нетто, г) указан в табл. 55.
Рассчитываем массовую долю сухих веществ в тесте исходя из пропорции:
|
6118 - 3803,7 |
|
|
|
100 - X |
Таблица 55
|
Сырье |
Расход сырья на 100 ватрушек массой по 75 г (нетто) |
Количество сухих веществ |
Количество жира |
Количество сахара |
||||
|
% |
г |
% |
г |
% |
г |
|||
|
Мука пшеничная высшего сорта |
3718 |
|
85,5* |
3178,89 |
- |
- |
- |
- |
|
Мука на подпыл |
174 |
148,77 |
- |
- |
- |
- |
||
|
Сахар |
197 |
99,86 |
196,72 |
|
|
99,8 |
196,61 |
|
|
Маргарин столовый (молочный) |
168 |
84,1 |
141,29 |
82,0 |
137,76 |
- |
- |
|
|
Меланж |
197 |
26,0 |
51,22 |
11,5 |
22,66 |
- |
- |
|
|
Соль |
58 |
99,8 |
57,88 |
- |
- |
- |
- |
|
|
Дрожжи (прессованные) |
110 |
26,0 |
28,6 |
- |
- |
- |
- |
|
|
Вода |
1496 |
- |
- |
- |
- |
- |
- |
|
|
Выход теста |
5800 |
|
|
|
|
|
|
|
|
Масса сырья |
6118 |
|
3803,37 |
- |
160,42 |
- |
196,61 |
|
_____________
* С учетом базисной влажности муки 14,5 %.
Влажность теста составит 100 - 62,17 % = 37,83 % или
![]()
Влажность основы ватрушек устанавливают путем анализа образцов, полученных при контрольных выпечках изделий, в количестве не менее трех.
В соответствии с рецептурой массовая доля жира в тесте и основе ватрушек в пересчете на сухое вещество составит:
![]()
При допустимом отклонении в меньшую сторону не более 0,5 % массовая доля жира в тесте и основе ватрушек должна быть не менее 3,7 %.
Массовая доля сахара в пересчете на сухое вещество составит:
![]()
При допустимом отклонении в меньшую сторону не более 1 % массовая доля сахара в тесте и основе ватрушек должна быть не менее 4,2 %.
Для изготовления 100 ватрушек используют фарш творожный в количестве 3 кг по рец. № 1135 (III колонка) Сборника рецептур блюд и кулинарных изделий, 1981 г. Расход сырья (нетто, г) указан в табл. 56.
Таблица 56
|
Сырье |
Расход сырья (нетто) на 3000 г фарша для 100 ватрушек, г |
Количество сухих веществ |
Количество сахара |
||
|
% |
г |
% |
г |
||
|
Творог полужирный |
2718 |
29,7 |
807,25 |
- |
- |
|
Яйца |
120 |
26,0 |
31,20 |
- |
- |
|
Сахар |
150 |
99,86 |
149,79 |
99,8 |
149,7 |
|
Мука пшеничная |
120 |
85,5 |
102,6 |
- |
- |
|
Ванилин |
0,3 |
- |
- |
- |
- |
|
Масса сырья |
3108,3 |
- |
1090,84 |
- |
149,7 |
|
Выход |
3000 |
- |
1052,83 |
- |
144,5 |
С целью проверки качества фарша на промежуточной стадии технологического процесса рассчитывают содержание в нем сухих веществ (X, %), исходя из следующей пропорции:
|
3000 - 1052,83 |
|
|
|
100 - X |
Массовая доля сахара в фарше творожном составит:
![]()
При допустимом отклонении в меньшую сторону не более 0,5 % массовая доля сахара в фарше должна быть не менее 4,3 %.
Содержание сахара в фарше готового изделия, а также фарша в процентах к массе изделия устанавливают путем анализа образцов, полученных при контрольных выпечках изделий, в количестве не менее трех.
Расчет физико-химических показателей - влажность теста, массовая доля сухих веществ фарша (полуфабриката), массовая доля сахара пирожков (печеных и жареных), пирогов, кулебяк производят аналогично расчету рецептуры ватрушки с творогом. При расчете минимально допустимого содержания сухих веществ фаршей, подвергавшихся тепловой обработке (мясной с луком, мясной с рисом, рисовый с яйцом, из капусты тушеной и т.п.), учитывают потери сухих веществ, равные 10 %, аналогично расчету сухих веществ для вторых блюд.
Пример расчета 2. Для изготовления 10 кг пирога полуоткрытого с использованием яблочного фарша, приготовленного по рец. № 1137 (1-й вариант) Сборника рецептур блюд и кулинарных изделий, 1981 г., требуется 4 кг фарша. Расход сырья (нетто, г) указан в табл. 57.
Таблица 57
|
Сырье |
Расход сырья (нетто) на 4000 г фарша, г |
Количество сухих веществ |
Количество сахара |
||
|
% |
г |
% |
г |
||
|
Яблоки свежие |
4048 |
13,0 |
526,24 |
9,0 |
364,32 |
|
Масса припущенных яблок |
3248 |
- |
- |
- |
- |
|
Сахар |
1200 |
99,86 |
1198,2 |
99,8 |
1197,6 |
|
Корица |
4 |
- |
4,0 |
- |
- |
|
Масса фарша после кулинарной обработки |
4452 |
- |
1728,44 |
- |
1561,92 |
|
Выход |
4000 |
- |
- |
- |
- |
Рассчитываем минимально допустимую массовую долю сухих веществ в фарше:
Хмин = 1728,44∙0,9 = 1555,6 г
Минимально допустимая массовая доля сухих веществ (Хмин, %) составит:
![]()
Массовая доля сахара в фарше составит:
![]()
При допустимом отклонении в меньшую сторону не более 2 % массовая доля сахара в фарше должна быть не менее 37 %.
Пример расчета 3. На анализ доставлен крем "Шарлотт", приготовленный по рец. № 39 (59) Сборника рецептур мучных кондитерских и булочных изделий для предприятий общественного питания, 1986 г. Согласно рец. № 39 в крем входит сироп "Шарлотт", рец. № 40 (60). Расход сырья (нетто, кг) сиропа "Шарлотт" указан в табл. 58.
Таблица 58
|
Сырье |
Содержание сухих веществ, % |
Расход сырья нетто, на 1 т, кг |
Сахар |
Жир |
|||
|
в натуре |
в сухих веществах |
% |
кг |
% |
кг |
||
|
Сахар |
99,85 |
631,34 |
630,39 |
99,7 |
629,44 |
- |
- |
|
Яйца |
27,00 |
112,24 |
30,30 |
- |
- |
10,0 |
11,22 |
|
Молоко цельное |
12,00 |
420,90 |
50,50 |
3,1* |
13,05 |
3,2 |
13,46 |
|
Масса сырья |
- |
1164,48 |
711,20 |
- |
642,49 |
- |
24,68 |
|
Выход |
68,5 |
1000,00 |
685,60 |
- |
619,38 |
- |
23,79 |
|
|
100,00 |
68,56 |
- |
61,94 |
- |
2,38 |
|
______________
* Лактоза молока выражена в сахарозе.
Примечание. Расчет массовой доли сухих веществ, жира и сахара произведен с использованием данных вышеуказанного Сборника.
Согласно рецептуре массовая доля общего сахара, выраженного в сахарозе (без пересчета на сухое вещество), составит:
![]()
(с допустимыми отклонениями по ТУ 10.04.08.13-88: минус 1,5, +2,0 %).
Массовая доля общего сахара (в пересчете на сухое вещество):
![]()
(с допустимыми отклонениями по ТУ 10.04.08.13-88: минус 1,5, +2,0 %).
Расход сырья (нетто, кг) крема "Шарлотт" (основанного на рецептуре № 39 (59) указан в табл. 59.
Таблица 59
|
Сырье |
Содержание сухих веществ, % |
Расход сырья нетто, на 1 т, кг |
Сахар |
Жир |
|||
|
в натуре |
в сухих веществах |
% |
кг |
% |
кг |
||
|
Масло сливочное |
84,00 |
422,23 |
354,68 |
- |
- |
82,50 |
348,34 |
|
Сироп "Шарлотт" № 40 (60) |
68,56 |
594,11 |
407,32 |
61,94 |
367,99 |
2,38 |
14,14 |
|
Пудра ванильная |
99,85 |
4,10 |
4,09 |
99,70 |
4,09 |
- |
- |
|
Коньяк или вино десертное |
- |
1,64 |
- |
- |
- |
- |
- |
|
Масса сырья |
|
1022,08 |
766,09 |
|
372,07 |
|
362,48 |
|
Выход |
75,00 |
1000,00 |
750,00 |
- |
363,77 |
- |
354,87 |
|
|
100,0 |
75,0 |
- |
36,38 |
- |
35,49 |
|
Влажность крема - 25 ± 2 %.
Массовая доля общего сахара, выраженного в сахарозе (без пересчета на сухое вещество), составит:
![]()
Массовая доля общего сахара в пересчете на сухое вещество:
|
|
с допускаемыми отклонениями |
48,6 - 1,5 = 47,1 % |
|
48,6 + 2 = 50,6 %. |
Массовая доля жира в пересчете на сухое вещество составит:
|
|
с допускаемыми отклонениями |
47,3 - 1,5 = 45,8 % |
|
47,3 + 2 = 49,3 % |
Рассчитываем водную фазу крема: в 100 г крема содержится 25 г воды и 36,4 г сахарозы, что в сумме составит 61,4 г (водная фаза).
Массовая доля сахарозы в пересчете на водную фазу составит:
![]()
Физико-химические показатели качества кремов представлены в табл. 60.
Таблица 60
|
Крем и № рецептуры Сборника 1986 г. |
Влажность, % |
Массовая доля жира в пересчете на сухое вещество, % |
Массовая доля общего сахара (сахарозы), % |
Массовая доля сухих веществ в водной вытяжке, по рефрактометру, % |
|
|
без пересчета на сухое вещество |
в пересчете на сухое вещество |
||||
|
Шарлотт № 39 (59) |
25 ± 2 |
47,3 |
36,4 |
48,5 |
40,0 |
|
Шарлотт шоколадный, № 45 (67) |
24,5 ± 2 |
43,6 |
35,6 |
47,2 |
40,0 |
|
Новый, № 41 (61) |
22 ± 2 |
48,6 |
38,7 |
49,6 |
46,6 |
7.4.2. Расчет содержания хлеба в полуфабрикатах и кулинарных изделиях из рубленого мяса по рецептуре (с учетом сухарной или пшеничной панировочной муки)
По ГОСТ 4288-76 "Изделия кулинарные и полуфабрикаты из рубленого мяса (Правила приемки и методы испытаний)" содержание хлеба в кулинарных изделиях из рубленого мяса должно определяться вместе с панировкой (сухарной панировочной мукой). Это требует пересчета на хлеб количества сухарной панировочной муки, предусмотренной по рецептуре.
Согласно ОСТ 49121-84 и рецептурам сборников для изготовления полуфабрикатов из рубленого мяса применяют хлеб и муку сухарную панировочную из хлеба, приготовленные из муки пшеничной не ниже I сорта.
Установлено, что среднее содержание углеводов (в процентах к массе) в пересчете на крахмал в хлебе из пшеничной муки I сорта при определении йодометрическим методом составляет 48,9 %, в сухарной панировочной муке - 73,7 %; при определении углеводов цианидным методом соответственно 46,2 и 67,1 %*(4).
Коэффициенты пересчета панировочной муки на хлеб соответственно составляют 1,5 и 1,45. Учитывая незначительную разницу между ними, при исследовании полуфабрикатов и кулинарных изделий из котлетной массы количество панировки по рецептуре пересчитывают на хлеб, используя коэффициент 1,5, а затем суммируют его с количеством хлеба по рецептуре. Далее вычисляют процентное содержание хлеба*(5).
________________
*(4) По данным исследований МИНХа им. Г.В. Плеханова.
*(5) При расчете содержания хлеба в тефтелях количество пшеничной муки I сорта пересчитывают на хлеб умножением на 1,5.
Пример расчета. По рец. № 658 (III колонка) Сборника 1981 г. в котлеты, биточки, шницели рубленые входят следующие компоненты сырья (нетто, г):
|
Говядина (котлетное мясо) |
37 |
|
|
Хлеб пшеничный |
9 |
|
|
Молоко или вода |
12 |
|
|
Сухари |
5 |
|
|
Масса полуфабриката |
62 |
|
|
Масса готового изделия |
50 |
|
1. Содержание хлеба в процентах в полуфабрикате (с учетом сухарной панировочной муки) составляет: 9 + 5∙1,5 = 16,5 г.
![]()
2. То же в готовом изделии:
![]()
Следовательно, содержание хлеба в полуфабрикате и готовом изделии не должно превышать соответственно 26,6 и 33 %.
Пример расчета. По рец. № 668 (III колонка) Сборника 1981 г. в тефтели (полуфабрикат) входят следующие виды сырья (нетто, г):
|
Говядина (котлетное мясо) |
38 |
|
|
Хлеб пшеничный I сорта |
8 |
|
|
Вода |
12 |
|
|
Лук репчатый пассерованный |
10 |
|
|
Мука пшеничная |
4 |
|
|
Масса полуфабриката |
71 |
|
Содержание хлеба по рецептуре, с учетом панировочной муки, рассчитываем, как указано выше:
![]()
Следовательно, согласно рецептуре содержание хлеба с учетом панировочной муки должно быть не более 19,7 %.
________________
*(6) При необходимости определения количества хлеба внутри готового изделия норму содержания хлеба устанавливают путем контрольного приготовления изделий с последующим проведением физико-химического анализа - в количестве не менее 5.
7.4.3. Расчет содержания муки и сахара в полуфабрикатах и изделиях из творога
Пример расчета. По рец. № 492 (I вариант) Сборника 1981 г. в сырники входит следующее сырье (нетто, г):
|
Творог |
135 |
|
|
Мука пшеничная |
20 |
|
|
Сахар |
15 |
|
|
Яйца |
5 |
|
|
Масса полуфабриката |
170 |
|
|
Выход готового изделия |
150 |
|
В соответствии с рецептурой, количество муки в сырниках не должно превышать 22 г (с учетом допустимого отклонения в большую сторону +10 %), или в процентах:
|
в полуфабрикате: |
|
|
в готовом изделии: |
|
Рассчитываем минимальное количество сахара (сахарозы) в полуфабрикате и готовом изделии (с учетом 3 % потерь):
![]()
14,97∙0,97 = 13,52 г, или 13,5 г.
Минимальное содержание сахара в сырниках (в процентах) составит:
|
в полуфабрикате: |
|
|
в готовом изделии: |
|
Аналогичным образом рассчитывают по рецептуре содержание риса в голубцах с мясом и рисом и в мясных фаршах с рисом; манной крупы - в муссах с манной крупой.
7.4.4. Расчет содержания молока (по результатам анализа)
Количество молока в блюдах и напитках определяют по содержанию лактозы*(7). Одновременно устанавливают массовую долю лактозы в молоке, используемом для приготовления этих блюд и напитков. Для этого при отборе пробы молочного блюда или напитка обязательно берут для анализа и пробу молока.
Количество молока (X, г) в порции блюда или напитка рассчитывают по формуле:
|
|
(105) |
где:
в - массовая доля лактозы в выпаренной части супа, порции каши или напитка, г;
a - фактическое содержание лактозы в молоке цельном или сгущенном, используемом для приготовления блюда, %.
Результат анализа сравнивают с количеством молока по рецептуре с учетом допустимых отклонений ±10 %.
Фактическое содержание молока (Х2, г) в молочном супе можно рассчитать также и по формуле*(8):
|
|
(106) |
где:
X - масса лактозы в молочном супе, %;
P - масса блюда, г;
X1 - массовая доля лактозы в молоке, определенная экспериментально, %;
K - коэффициент пересчета, учитывающий углеводы гарниров, равен 0,790 при определении лактозы в жидкой части и 0,803 - в гомогенизированном супе.
Этот коэффициент установлен путем деления теоретического содержания лактозы в блюде на фактическое, установленное экспериментально одним из методов определения сахаров, в супе, приготовленном строго по рецептуре (не менее 3-х опытов).
Содержание лактозы в супе целесообразно определять в жидкой части или гомогенизированном супе, исключив при подготовке проб молочных супов упаривание.
_________________
*(7) Массовая доля лактозы в молоке, блюде (напитке) должна быть определена одним и тем же методом. При определении лактозы перманганатным методом пользуются табл. 14.
*(8) Разработаны лабораторией ХИОПа.
7.4.5. Определение химического состава и энергетической ценности (калорийности) пищи
Химический состав кулинарной продукции определяют с целью проверки соответствия его рекомендуемым нормам потребности в пищевых веществах, а также для подсчета энергетической ценности пищи. Как правило, определяют химический состав рационов или отдельных приемов пищи для учащихся профтехучилищ, студентов, школьников, отдыхающих в здравницах и др., а также состав скомплектованных обедов (завтраков, ужинов).
Пользуясь справочными таблицами содержания основных пищевых веществ и энергетической ценности пищевых продуктов, рассчитывают химический состав всех продуктов, входящих в рецептуру блюда. Данные по содержанию в каждом продукте белков, углеводов и жира суммируют. Суммарное количество белков, углеводов и жира блюда (рациона) умножают на соответствующие коэффициенты энергетической ценности (табл. 61), учитывающие только усвояемую энергию пищевых веществ.
Таблица 61
Коэффициенты энергетической ценности пищевых веществ
|
Пищевые вещества |
Коэффициент, ккал/г |
|
Белки |
4,0 |
|
Жиры |
9,0 |
|
Углеводы "по разности"* |
4,0 |
|
Сумма моно- и дисахаридов |
3,8 |
|
Крахмал, определенный экспериментально |
4,1 |
|
Клетчатка |
0,0 |
|
Органические кислоты** |
|
|
уксусная |
3,5 |
|
яблочная |
2,4 |
|
молочная |
3,6 |
|
лимонная |
2,5 |
_________________
* Для определения углеводов "по разности" из сухого остатка продукта или блюда вычитают количество белка, жиров и золы.
** Если кислота неизвестна или имеется смесь кислот, то используют коэффициент 3,0.
Энергетическая ценность отдельного приема пищи или рациона (X, ккал) равна сумме этих произведений, т.е.
где 4,0; 4,0; 9,0 - коэффициенты энергетической ценности соответственно белков, углеводов и жиров, ккал/г;
Б, У, Ж - количество соответственно белков, углеводов, жира в блюде (приеме пищи, рационе), г.
В случае необходимости выразить энергетическую ценность в килоджоулях, полученное число килокалорий умножают на 4,184.
Расчетные данные сравнивают с "Нормами физиологических потребностей в пищевых веществах и энергии для различных групп населения СССР", утвержденными Минздравом СССР в 1991 г.
В справочных Таблицах представлен химический состав продуктов, не прошедших в большинстве своем тепловую обработку. Последняя же, как известно, сопровождается потерей части сухих веществ (белков, жиров, углеводов). Чтобы рассчитать энергетическую ценность блюд с учетом этих потерь, пользуются справочными таблицами содержания основных пищевых веществ и энергетической ценности блюд и кулинарных изделий (том III).
При отсутствии в таблицах необходимого блюда или изделия химический состав его рассчитывают следующим образом: определяют химический состав сырьевого набора изделия, пользуясь таблицами справочника "Химический состав пищевых продуктов" (тт. I - III); находят размер потерь отдельных сухих веществ при аналогичном способе тепловой обработки (варке, жарке и т.д.) основного и дополнительного продуктов в III томе того же справочника и определяют выход готового изделия как отношение его массы (по рецептуре) к массе исходного сырьевого набора (в %).
Массу белков, жиров и углеводов в изделии (с учетом их потерь при тепловой обработке) вычисляют в мг или г на 100 г съедобной части (Kг) по формуле:
|
|
(108) |
где:
Св - сохранность вещества, определяемая путем вычитания процента потерь на 100;
Kп - содержание исследуемого пищевого вещества в 100 г съедобной части сырьевого набора, мг или г;
М - выход готового изделия, определяемый как отношение его массы Мг к массе сырьевого набора Мп, %.
Пример расчета. Рец. № 605 изделия "Колбаса жареная по-ленинградски": колбаса вареная 55 г, мука 3, яйца 4, сухари 10, маргарин столовый 6 г. Масса набора сырья 78 г. Масса готового изделия 60 г.
Содержание белка в сырьевом наборе: 6,7 + 0,32 + 0,51 + 1,12 + 0,02 = 8,67, или в пересчете на 100 г сырьевого набора 11,1 г.
Если потери белка при жарке - 10 %, сохранность = 90 %. Выход изделия 60∙100:78 = 77 %. Содержание белка в пересчете на 100 г готового изделия: 90∙11,1:77 = 12,9, т.е. в изделии массой 60 г 12,9∙60:100 = 7,74 г.
Так же определяют количество жира и углеводов в изделии. Умножив найденные количества белков, жиров и углеводов на соответствующие энергетические коэффициенты, суммируют полученные величины и получают физиологическую энергетическую ценность готового изделия.
Как известно, энергетическая ценность может остаться неизменной при замене одного продукта другим, например белкосодержащего продукта - продуктом, богатым углеводами. Поэтому наряду с расчетным определением химического состава практикуется лабораторный анализ пищи на содержание сухих веществ, белков и жира.
Энергетическую ценность рациона (X, ккал) определяют по формуле:
|
X = ∕C - (Б + Ж + M)∕4,0 + 4,0Б + 9,0Ж, |
(109) |
где М - содержание минеральных веществ, г;
С - содержание сухих веществ, г.
Остальные обозначения те же, что в формуле (107).
В отличие от расчетной, энергетическую ценность, вычисленную на основании данных анализа, называют фактической.
Фактическую энергетическую ценность сравнивают с расчетной минимально допустимой, подсчитанной с учетом потерь сухих веществ, жиров и белков при приготовлении и порционировании блюд.
Если фактическая энергетическая ценность суточного рациона ниже расчетной минимально допустимой, значит, имеет место недовложение сырья в блюда. Если же она значительно превышает минимально допустимую, то определяют максимальную энергетическую ценность. В этом случае расчет количества сухих веществ (в том числе белков и жиров) ведут без учета потерь. При этом учитывают жир, содержащийся в крупе, муке и других продуктах растительного происхождения. Превышение фактической энергетической ценности над расчетной максимальной указывает на нарушение норм вложения сырья.
При выдаче результатов анализов обязательно указывают и отклонения фактических данных от расчетных по содержанию белков, углеводов и жиров.
Раздел 8. Методы определения правильности использования синтетических красителей и осуществление санитарного контроля
8.1. Методы определения правильности использования синтетических красителей
Согласно циркулярному письму Министерства торговли СССР № 0135-75 от 23 июня 1969 г. и № 039-75 от 17 февраля 1967 г. запрещено применение в общественном питании для подкрашивания продукции импортного амаранта и производство отечественной амарантной пасты.
Для подкрашивания кондитерских изделий, холодных первых и сладких блюд можно применять безвредные красители, разрешенные органами санитарно-эпидемиологической службы Министерства здравоохранения СССР.
В основном это естественные красители - соки съедобных плодов, ягод, овощей и продукты их переработки, растительные красители из чая, настой кофе, порошок какао, жженый сахар, шафран, кармин и т.д.
Из искусственных красителей разрешается применять индигокармин и тартразин.
8.1.1. Обнаружение красного синтетического красителя (амаранта, анилинового красителя) в сиропах, компотах, напитках, водных растворах красителей и кондитерских кремах
Метод основан на способности раствора аммиака изменять красный цвет натуральных и оставлять без изменения цвет синтетических пищевых красителей (анилиновых, амаранта).
Аппаратура, материалы, реактивы. Весы лабораторные; пробирки; штатив для пробирок; баня водяная; капельница стеклянная лабораторная; колба мерная вместимостью 50 см3; чашка фарфоровая; пипетка вместимостью 1 и 20 см3; палочка стеклянная; гидроксид аммония, раствор с массовой долей 10 % (20 мл водного аммиака массовой долей 25 % переносят в мерную колбу на 50 см3 и объем доводят до метки водой); раствор амаранта (0,015 г амаранта растворяют в 1 дм3 дистиллированной воды); вода дистиллированная.
Проведение испытания. В пробирку отбирают 3 см3 испытуемого раствора, добавляют 4 капли раствора аммиака с массовой долей 10 % и встряхивают. Наблюдения ведут через 1 - 2 мин. Если в растворе содержится натуральный краситель, то красный цвет исчезает и раствор приобретает темную окраску с зеленоватым оттенком. При наличии в испытуемом растворе синтетического красителя (амаранта, анилинового красителя) цвет его не изменяется.
В случае необходимости смесь растворов может быть оставлена на сутки и больше. Это не отражается на результатах реакции. При наличии синтетического красителя раствор становится более прозрачным, а цвет более ярким.
При проведении исследований непосредственно на предприятии желательно вести сравнение с раствором амаранта 0,015 г/дм3.
При исследовании окрашенного кондитерского крема готовят водную вытяжку: 2 - 3 г крема тщательно размешивают в фарфоровой чашке, добавляют 8 - 10 см3 воды, после перемешивания ставят на кипящую водяную баню и нагревают до температуры плавления жира.
Полученный раствор быстро охлаждают, поместив его в холодильник, затем снимают жир, который соберется на его поверхности. Далее проводят анализ раствора, как указано выше.
Обнаружение амаранта. Метод основан на способности раствора сернокислой меди в присутствии уксусной кислоты изменять цвет исследуемого раствора при наличии в нем амаранта.
Аппаратура, материалы, реактивы. Пробирки, штатив для пробирок; пипетка вместимостью 5 см3; сульфат меди (II), раствор с массовой долей 1 %; кислота уксусная.
Проведение испытания. К 5 см3 исследуемого раствора добавляют 1 см3 раствора сульфата меди (II). При наличии амаранта раствор приобретает желтую окраску, переходящую в розовую при добавлении нескольких капель уксусной кислоты.
При исследовании окрашенного кондитерского крема готовят водную вытяжку, как указано выше, и проводят реакцию с сульфатом меди.
8.1.2. Метод идентификации разных синтетических и натуральных пищевых красителей
Метод основан на способности синтетических красителей удерживаться на шерстяной нити.
Аппаратура, материалы, реактивы. Нить шерстяная белая; баня водяная; часы песочные на 10 мин.; штатив для пробирок; пробирки; мыло хозяйственное.
Проведение испытания. В пробирку с раствором красителя погружают кусочек белой обезжиренной шерстяной нити длиной 2 - 3 см, после чего пробирку помещают в кипящую водяную баню на 10 мин. Кусочек нити окрашивается в цвет, характерный для данного красителя. По истечении 10 мин. кусочек пряжи извлекают и тщательно промывают проточной водой с мылом.
Если нить окрашена синтетическим красителем, то цвет после отмывания не меняется. При окрашивании натуральным красителем (соком свеклы, моркови, черной смородины, клюквы и т.д.) он легко смывается или пряжа после отмывания приобретает грязно-бурый оттенок.
8.2. Простейшие инструментальные и химические методы обследования при осуществлении санитарного контроля
Контроль осуществляется санитарно-технологическими пищевыми лабораториями.
8.2.1. Контроль за соблюдением температуры воды при обработке посуды
Режим мытья столовой посуды предусмотрен действующими "Санитарными правилами для предприятий общественного питания". Нарушение температурного режима должно рассматриваться как одно из самых грубых отступлений от санитарных правил.
Измерение температуры воды в ваннах следует проводить термометром со шкалой до 100 °C в момент наибольшей нагрузки предприятия (во время обеда) 5 раз в течение получаса или 10 раз в течение часа, то есть через каждые 6 мин. - время, в течение которого обычно моется одна партия посуды. Первое измерение производят внезапно, без предупреждения. Это дает возможность зафиксировать температурные условия, при которых обрабатывается посуда.
Результаты измерения температуры вносят в акт обследования или фиксируют его на специальном бланке (см. схему).
|
"__" _______________ 19___ г. (дата) Столовая, ресторан, кафе, буфет № _______________ (подчеркнуть) Результаты измерения температуры воды для мытья столовой посуды
Замеры температуры воды произведены в течение ______ с интервалом в _____ мин. Мытье посуды осуществляется ручным способом, машиной (нужное подчеркнуть) Горячая вода подается из водопровода, из титана, подогревается на плите (нужное подчеркнуть) Проверил _____________________________ Копию получил ________________________ Директор (столовой, ресторана ...) ________________________________
|
8.2.2. Определение концентрации щелочи в воде моечных ванн (обезжиривание посуды в первой моечной ванне)
Аппаратура, материалы, реактивы. Пробирка химическая диаметром 20 мм, высотой около 19 см; пипетка вместимостью 10 см3; капельница стеклянная лабораторная; фенолфталеин, спиртовой раствор с массовой долей 1 %; кислота соляная концентрацией 0,1 моль/дм3; карбонат натрия, раствор с массовой долей 5 %.
Проведение испытания. Определение концентрации щелочи*(9) в воде моечных ванн проводят с помощью специальной градуированной пробирки, рис. 6.
Рис. 6. Градуированная пробирка для определения
концентрации моющих средств
_____________
*(9) Минимально допустимая концентрация 0,5 %.
Градуировка пробирки. В пробирку (рис. 6) наливают 10 см3 раствора соды (Na2CO3) с массовой долей 5 % и добавляют 2 капли раствора фенолфталеина с массовой долей 1 %. На уровне этой жидкости наносят первую круговую метку "А". Затем постепенно (каплями) добавляют 0,1 моль/дм3 раствор соляной кислоты.
После каждого добавления жидкость перемешивают. Когда жидкость в пробирке обесцветится, отмечают ее уровень путем нанесения метки "Б".
В пробирку до нижней метки "А" наливают исследуемую воду (10 см3) и добавляют две капли раствора фенолфталеина с массовой долей 1 %. Щелочная жидкость приобретает розово-красный цвет. После этого постепенно (по каплям) добавляют 0,1 моль/дм3 раствора соляной кислоты. При каждом добавлении содержимое пробирки перемешивают. Если жидкость обесцветилась при добавлении кислоты ниже метки "Б", то концентрация щелочи в моечной ванне меньше нижней границы нормы (0,5 %).
Если обесцвечивание произошло на уровне метки "Б" и выше, то она в пределах нормы.
8.2.3. Проба на обнаружение хлора в моечных ваннах
В тех случаях, когда для обеззараживания посуды применяют хлорсодержащие препараты (хлорную известь, хлорамин), возникает необходимость контроля за правильностью их применения. Делают это с помощью индикаторной бумаги, пропитанной раствором йодидокалиевого крахмала.
Проведение испытания. Индикаторную бумагу, пропитанную раствором йодидокалиевого крахмала (см. раздел 9, п. 9.6.11) смачивают водой из второй моечной ванны, в которую входят хлорсодержащие препараты. При наличии хлора в воде бумага становится темно-синей. При смачивании обычной водопроводной водой цвет бумаги не меняется.
8.2.4. Определение качества мытья столовой посуды
Метод М.М. Балашова
Метод основан на окрашивании реактивом, содержащим краску судан III.
Проведение испытания. В тарелку наливают 5 см3 реактива (см. раздел 9, п. 9.6.12), содержащего краску судан III, и распределяют его по всей поверхности тарелки. Через 10 с на плохо обезжиренных тарелках образуются желтые пятна разной интенсивности. Различают три степени чистоты посуды: первая -поверхность посуды не желтеет, вторая - появляются отдельные пятна или полосы, третья - вся поверхность посуды окрашивается в желтый цвет.
Метод М.П. Болотова
Метод основан на окрашивании жира специальным реактивом, содержащим краску судан III.
Проведение испытания. Тарелку протирают полоской фильтровальной бумаги размером 15×15 мм, которую затем смачивают несколькими каплями специального реактива (см. раздел 9, п. 9.6.13) и через 10 с промывают холодной водой. При наличии на тарелке жира бумага окрашивается в желтый цвет, при отсутствии - в сероватый или голубоватый. Метод позволяет определить 0,2 - 0,5 мг жира на шероховатой поверхности пластмассовой тарелки.
Допускается на пищевом объекте брать только "бумажные пробы", а окрашивание их производить в лаборатории.
Метод А.К. Кощеева
Метод основан на определении количества жира, оставшегося на посуде, с помощью полосок из белой хлопчатобумажной ткани, смоченных эфиром.
Аппаратура, материалы, реактивы. Полоски белой хлопчатобумажной ткани, эфир этиловый; метиленовый голубой, раствор с массовой долей 0,01 %.
Проведение испытания. Полоской (рис. 7), смоченной эфиром и укрепленной на основании корковой пробки, протирают поверхность тарелки. После высушивания на воздухе полоску окрашивают. Для этого ее осторожно кладут на поверхность раствора метиленового голубого с массовой долей 0,01 % так, чтобы она смачивалась только с нижней стороны. При наличии жира на полоске остается круглое неокрашенное пятно, имеющее форму основания пробки. При отсутствии жира полоска окрашивается в синий цвет равномерно (рис. 7а). По величине неокрашенной части полоски можно судить о степени загрязнения посуды. Чем больше жира осталось на посуде, тем отчетливее и больше неокрашенное пятно.
Рис. 7. Полоски для определения степени чистоты столовой посуды
а - посуда чистая; б - посуда грязная
При отсутствии белого пятна посуду следует считать чистой и записывать в протоколе (акте) "обезжирена" или обозначать знаком минус (-).
Наличие на полоске отчетливого светлого пятна величиной с 1 - 2 спичечные головки или слабо заметного пятна неполного круга свидетельствует о незначительном загрязнении посуды (первая степень) и обозначается знаком "+". Если светлое пятно достигает 1/3 или 1/2 размера круга, загрязнение посуды относится ко второй степени и обозначается знаком "++". При третьей степени загрязнения на полоске отчетливо выявляется светлое пятно полного диаметра основания пробки, что обозначается знаком "+++".
Метиленовый голубой может быть заменен обычными анилиновыми чернилами в соответствующей концентрации (0,01 %). Вместо хлопчатобумажных полосок можно воспользоваться фильтровальной бумагой, однако чувствительность и точность метода при этом значительно снижается.
8.2.5. Контроль качества мытья столовой посуды после обработки ее водой с хлорсодержащими препаратами
При проведении по тарелке ватным тампоном, смоченным раствором йодистокалиевого крахмала, появляется полоса буровато-синего цвета. Если тарелки вымыты без применения хлорной извести, то цветная полоса не появляется.
8.2.6. Определение качества мытья вилок
Аппаратура, материалы, реактивы. Полоски хлопчатобумажные; ватные тампончики на спичках; лупа; эфир этиловый.
Проведение испытания. Качество мытья вилок можно определять так же, как и тарелок, протирая их хлопчатобумажными полосками, смоченными эфиром (см. с. 265). Кроме того, для обнаружения остатков пищи между зубцами можно использовать лупу и ватные тампончики на спичках. Тампончиком протирают вилку между зубцами и затем рассматривают прилипшие к нему частички пищи в лупу. Для проверки следует брать не менее 10 вилок. При обнаружении хотя бы одной частички пищи вилки считаются плохо вымытыми.
8.2.7. Определение количества активного хлора, содержащегося в порошке хлорной извести
Аппаратура, материалы, реактивы. Весы лабораторные; бюкса стеклянная; колба коническая вместимостью 200 см3 с притертой пробкой; цилиндр измерительный вместимостью 50 см3; бюретка вместимостью 50 см3; пипетка вместимостью 2 см3; кислота серная, раствор с массовой долей 20 %; йодид калия, раствор с массовой долей 10 %; тиосульфат натрия, раствор 0,1 моль/дм3; крахмал растворимый, раствор массовой долей 1 %; вода дистиллированная.
Проведение испытания. Навеску порошка хлорной извести около 2 г (точность до 0,001 г) в стеклянной бюксе количественно переносят в коническую колбу вместимостью 200 см3 с притертой пробкой, при трехкратном ополаскивании бюксы заранее отмеренными 50 см3 дистиллированной воды.
Содержимое колбы перемешивают в течение 1 мин, затем добавляют 20 см3 раствора серной кислоты с массовой долей 20 % и 20 см3 раствора йодида калия с массовой долей 10 %.
Колбу закрывают пробкой, осторожно взбалтывают и ставят на 5 мин в темное место. Выделившийся йод титруют раствором тиосульфата натрия с концентрацией 0,1 моль/дм3 (0,1 н) до соломенно-желтого цвета. После этого добавляют 1 см3 раствора крахмала с массовой долей 1 % и дотитровывают до полного обесцвечивания раствора.
Обработка результатов. Содержание активного хлора (X, %) рассчитывают по формуле:
|
|
(110) |
где:
V - объем 0,1 моль/дм3 раствора тиосульфата натрия, пошедшего на титрование, см3;
K - поправочный коэффициент к титру 0,1 моль/дм3 раствора тиосульфата натрия;
m - навеска порошка хлорной извести, г;
0,003546 - количество хлора, соответствующее 1 см3 0,1 моль/дм3 раствора тиосульфата натрия, г.
Пример расчета:
V = 1400 см3; K = 0,9804; m = 2,0 г;
![]()
По Санитарным правилам, утвержденным Министерством здравоохранения СССР, в воде второй моечной ванны должно содержаться 0,2 % хлорной извести или 0,2 % хлорамина. Для этого необходимо вносить 200 см3 осветленного раствора хлорной извести с массовой долей 10 % или 20 г хлорамина на 10 л воды (1 ведро).
Обычно готовят раствор хлорной извести с массовой долей 10 %, а затем осветленный раствор разводят, как указано выше, и получают требуемое содержание хлорной извести.
Раздел 9. Приготовление растворов реактивов
9.1. Растворы кислот
9.1.1. Серная кислота. Исходным раствором является концентрированная серная кислота (Н2SO4), х.ч., по ГОСТ 4204-77, плотностью 1,84 г/см3. Плотность кислоты необходимо проверять с помощью ареометра.
2 моль/дм3 (4 н) раствор. Отмеряют мерным цилиндром III см3 концентрированной серной кислоты. Осторожно, небольшими порциями, при охлаждении, приливают ее к дистиллированной воде. После охлаждения объем доводят до 1000 см3.
0,5 моль/дм3 (1 н) раствор. Отмеривают мерным цилиндром 27,8 см3 концентрированной серной кислоты и разбавляют дистиллированной водой до 1000 см3, как указано выше.
0,05 моль/дм3 (0,1 н) раствор. Для его приготовления 2 моль/дм3 (4 н) или 0,5 моль/дм3 (1 н) растворы разбавляют, соответственно, в 40 или 10 раз: 1 объем 2 моль/дм3 (4 н) раствора кислоты и 39 объемов дистиллированной воды или 1 объем 0,5 моль/дм3 (1 н) раствора кислоты и 9 объемов дистиллированной воды. Полученный раствор перемешивают и не ранее чем на следующий день устанавливают коэффициент поправки.
Для определения коэффициента поправки берут несколько отдельных навесок по 0,15 - 0,2 г подсушенного при 150 °C карбоната натрия (натрия углекислого безводного), х.ч., с точностью до 0,0002 г. Их количественно переносят с помощью дистиллированной воды в коническую колбу вместимостью 250 см3 так, чтобы объем стал около 25 см3. Затем добавляют 1 - 2 капли раствора метилового оранжевого и титруют из бюретки приготовленным раствором кислоты до перехода желтой окраски в оранжево-розовую. Для определения коэффициента поправки можно использовать тетраборат натрия (буру); навеску берут массой около 0,5 г растворяют и переносят ее в коническую колбу с помощью теплой дистиллированной воды в количестве 30 - 60 см3. Добиваются полного растворения навески. В качестве индикатора применяют раствор метиленового красного с массовой долей 1 %. Затем раствор титруют, как и при использовании карбоната натрия.
Коэффициент поправки в обоих случаях вычисляют по формуле:
|
|
(111) |
где:
K - коэффициент поправки;
V - израсходованный на титрование объем раствора, в котором устанавливает коэффициент поправки, см3;
a - навеска исходного вещества, г;
σ - количество исходного вещества, соответствующее 1 см3 0,1 моль/дм3 раствора кислоты, г (для карбоната натрия а = 0,0053 г, для тетрабората натрия σ = 0,019072 г).
0,05 моль/дм3 (0,1 н) раствор кислоты удобно готовить из фиксанала; в этом случае коэффициент поправки не устанавливают.
Коэффициент поправки 0,5 моль/дм3 (1 н) раствора серной кислоты устанавливают так, как указано для 0,05 моль/дм3 (0,1 н) раствора; при этом количество карбоната натрия и тетрабората натрия берут в 10 раз больше; тогда величина σ будет соответственно: для карбоната натрия - 0,053 г, для тетрабората - 0,19072 г.
0,01 моль/дм3 (0,02 н) раствор. Пипеткой с грушей отмеривают 0,56 см3 концентрированной серной кислоты и приливают ее к дистиллированной воде, затем объем доводят до 1000 см3.
Раствор с массовой долей 25 %. Один объем концентрированной серной кислоты смешивают с пятью объемами воды.
Раствор с массовой долей 10 %. Отмеривают цилиндром 59 см3 концентрированной серной кислоты и приливают к 940 см3 дистиллированной воды.
Кислота серная 1:2 по объему. Отмеривают цилиндром необходимый объем концентрированной серной кислоты и осторожно малыми порциями (при охлаждении) приливают к двухкратному объему дистиллированной воды.
9.1.2. Соляная кислота. Исходным раствором является концентрированная соляная кислота (HCl), х.ч., по ГОСТ 3118-77, плотностью 1,19 г/см3, которую проверяют ареометром.
1 моль/дм3 (1 н) раствор. Отмеривают мерным цилиндром 82,2 см3 концентрированной соляной кислоты и растворяют ее в дистиллированной воде. Объем доводят до 1000 см3, тщательно перемешивают и не ранее чем на следующий день устанавливают коэффициент поправки так, как указано в п. 9.1.1 для 0,5 моль/дм3 (1 н) раствора серной кислоты.
0,5 моль/дм3 (0,5 н) раствор. В 2 раза разбавляют 1 моль/дм3 (1 н) раствор соляной кислоты водой, то есть 1 объем 1 моль/дм3 (1 н) раствора и 1 объем воды.
0,1 моль/дм3 (0,1 н) раствор. В 10 раз разбавляют 1 моль/дм3 (1 н) раствор соляной кислоты водой, то есть 1 объем 1 моль/дм3 (1 н) и 9 объемов воды. Полученный раствор тщательно перемешивают и не ранее чем на следующий день устанавливают коэффициент поправки так, как указано для 0,05 моль/дм3 (0,1 н) раствора серной кислоты (п. 9.1.1).
0,1 моль/дм3 (0,1 н) раствор соляной кислоты удобно готовить из фиксанала без установления коэффициента поправки.
Растворы с массовой долей 20 %, 10 % и 2 % (приблизительно) готовят путем разбавления, соответственно 483, 231 и 45 см3 концентрированной соляной кислоты дистиллированной водой до объема 1000 см3.
9.1.3. Азотная кислота. Исходным реактивом является концентрированная азотная кислота (HNO3), х.ч., по ГОСТ 4461-77, плотностью 1,4 г/см3, которую проверяют ареометром.
Растворы с массовой долей 20 % и 10 % (приблизительно) готовят разбавлением, соответственно: 233 и 111 см3 концентрированной азотной кислоты дистиллированной водой до объема 1000 см3, а затем тщательно перемешивают.
9.1.4. Уксусная кислота. Исходным реактивом является концентрированная уксусная кислота (C2H4О2), х.ч., по ГОСТ 61-75, с массовой долей 98 % (ледяная) или кислота уксусная лесохимическая по ГОСТ 6968-76 (раствор с массовой долей 80 %).
Раствор с массовой долей 12 %. 11,6 см3 ледяной уксусной кислоты растворяют в 88 см3 дистиллированной воды. Если для приготовления раствора используют уксусную кислоту с массовой долей 80 % (плотностью 1,07 г/см3), ее отмеривают в количестве 14 см3 и растворяют в 86 см3 воды.
9.1.5. Кислота трихлоруксусная (CCl3COOH), х.ч., по ТУ 6-09-1926-72, растворы с массовой долей 30 % и 40 %. Соответственно 300 г и 400 г кислоты трихлоруксусной растворяют в 700, 600 см3 дистиллированной воды.
9.2. Растворы щелочей
9.2.1. Гидроксиды натрия или калия. Исходными реактивами являются гидроксид натрия (едкий натр), NaOH, х.ч. или ч.д.а., по ГОСТ 4328-77 или гидроксид калия (едкое кали), KOH, х.ч. или ч.д.а., по действующей нормативно-технической документации.
2,5 моль/дм3 (2,5 н) раствор. Вначале готовят концентрированный раствор с массовой долей примерно 45 %. Для этого в фарфоровой посуде взвешивают на технических весах 500 - 520 г NaOH или KOH, смывают водой верхний слой карбоната. Обмытые гранулы растворяют в 500 см3 воды, добавляя ее постепенно, при непрерывном помешивании. Остывший концентрированный раствор осторожно сливают в бутыль, которую закрывают пробкой с хлоркальциевой трубкой, наполненной натронной известью*(10). Раствор оставляют для отстаивания на срок не менее двух дней, затем сливают с осадка и замеряют плотность ареометром.
______________
*(10) Все растворы гидроксида натрия или калия хранят, защищая их от углекислого газа воздуха при помощи вставленных в пробки хлоркальциевых трубок, заполненных натронной известью. Натронную известь получают при взаимодействии концентрированного раствора NaOH со свежепрокаленной CaO (на 2 весовые части CaO одна весовая часть NaOH) с последующим выпариванием (осторожно) и слабым прокаливанием. После измельчения и просеивания ею заполняют хлоркальциевую трубку.
Из концентрированного раствора путем разбавления водой (свободной от СО2) готовят приблизительно 2,5 моль/дм3 раствор с массовой долей 10 %, плотностью 1,109 г/см3. Для удаления углекислого газа дистиллированную воду кипятят и охлаждают в колбе с закрытой пробкой, в которую вставлена хлоркальциевая трубка, заполненная натронной известью.
Точность 2,5 моль/дм3 раствора проверяют титрованием соляной или серной кислотой в присутствии фенолфталеина.
На 10 см3 точно 2,5 моль/дм3 (2,5 н) раствора гидроксида натрия или гидроксида калия должно пойти 25 см3 точно 1 моль/дм3 (1 н) раствора соляной или 0,5 моль/дм3 (1 н) раствора серной кислоты. Если кислоты идет на титрование больше или меньше, то концентрацию раствора гидроокиси натрия соответственно уменьшают, разбавляя водой, или увеличивают, добавляя раствор гидроксида натрия с массовой долей 45 %.
1 моль/дм3 (1 н) раствор. Из концентрированного раствора путем разбавления водой, освобожденной от , готовят раствор с массовой долей 4 % (приблизительно), плотностью 1,043 г/см3.
Коэффициент поправки 1 моль/дм3 растворов NaOH и KOH устанавливают по 1 моль/дм3 (1 н) раствору соляной или 0,5 моль/дм3 (1 н) серной кислоты, приготовленному из фиксанала (К = 1), или с установленным коэффициентом поправки. В этом случае в 3 - 4 конические колбы отмеривают пипеткой 10 - 20 см3 растворов NaOH или KOH добавляют по 3 - 4 капли раствора фенолфталеина с массовой долей 1 % и титруют соответствующим раствором соляной или серной кислоты до исчезновения розовой окраски. Коэффициент поправки вычисляют по формуле:
|
|
(112) |
где:
K - коэффициент поправки;
V1 - объем раствора кислоты, израсходованной на титрование, см3;
K1 - коэффициент поправки использованного раствора кислоты;
V2 - объем раствора NaOH или KOH, взятый для титрования, см3.
Коэффициент поправки растворов NaOH и KOH проверяют не реже 1 раза в 3 месяца, а при наличии резких колебаний температуры окружающего воздуха - чаще.
1 моль/дм3 (1 н) раствор, используемый для осаждения мешающих несахаров в комплексе с 0,5 моль/дм3 (1 н) раствором сульфата цинка, может содержать некоторое количество карбонатов и при этом хорошо выполнять свою функцию. Его можно приготовить непосредственно из сухого реактива: взвешивают 40 - 45 г NaOH или 56 - 60 г KOH в фарфоровой чашке, смывают небольшим количеством воды без CO2 верхний слой карбоната, обмытые кристаллы растворяют в 100 - 150 см3 воды и, после охлаждения, разбавляют до объема 1000 см3. Затем устанавливают эквивалентное соотношение этих растворов, как указано ниже.
0,1 моль/дм3 (0,1 н) раствор. Приготовленный 1 моль/дм3 (1 н) раствор NaOH разбавляют в 10 раз дистиллированной водой, освобожденной от CO2. Один объем 1 моль/дм3 (1 н) раствора NaOH и 9 объемов дистиллированной воды смешивают непосредственно в сосуде для хранения раствора, а затем устанавливают коэффициент поправки.
Растворы с массовой долей 15 % и 2 % (приблизительно). Взвешивают на технических весах соответственно 150 и 20 г твердого NaOH, осторожно растворяют в небольшом количестве дистиллированной воды, освобожденной от CO2, и разбавляют такой же водой до 1000 см3.
Если приготовлен предварительно концентрированный раствор NaOH, то его разбавляют дистиллированной водой без CO2 до плотности соответственно 1,164 и 1,021 г/см3.
9.2.2. Гидроксид бария, насыщенный раствор. Исходным реактивом является гидроксид бария (Ba(OH)2∙8Н2O), х.ч. или ч.д.а., по ГОСТ 4107-78. Растворяют в воде Ba(OH)2 при нагревании (70 - 80 °C) до насыщения. При охлаждении из раствора выпадает кристаллический гидроксид бария с большим содержанием в нем карбоната бария; прозрачную жидкость осторожно сливают с помощью сифона в склянку, из которой предварительно удаляют углекислоту, пропуская через нее поток воздуха, лишенного углекислоты (в течение нескольких часов), для чего воздух пропускают через промывные склянки с концентрированным раствором едкого кали или едкого натра или V-образные трубки с натронной известью.
С этой целью склянку, из которой удаляют углекислоту, соединяют с промывными склянками с помощью стеклянной трубки, вставленной в пробку, промывные склянки также соединяют между собой с помощью стеклянных трубок, вставленных в пробки (рис. 8). Вторую стеклянную трубку склянки присоединяют к водоструйному насосу посредством резиновой трубки с зажимом. По окончании продувания резиновую трубку перекрывают зажимом.
Рис. 8. Приспособление для очистки воздуха от углекислого газа
1 - промывные склянки с концентрированным раствором NaOH;
2 - склянка, из которой удаляют углекислоту;
3 - патрубок для соединения с вакуумом-насосом
9.2.3. Гидроксид аммония (водный раствор аммиака с массовой долей 25 %), х.ч. или ч.д.а., по ГОСТ 3760-79, растворы с массовой долей 15 % и 10 %. Готовят разбавлением соответственно 622 и 423 см3 водного раствора с массовой долей 25 % аммиака дистиллированной водой до 1000 см3.
9.3. Растворы солей
9.3.1. Сульфат цинка. Исходным реактивом является сульфат цинка (ZnSO4∙7Н2O), х.ч. или ч.д.а., по ГОСТ 4174-77.
0,5 моль/дм3 (1 н) раствор. 145 г реактива растворяют в дистиллированной воде в колбе вместимостью 1000 см3, доводят до метки и тщательно перемешивают.
Для применения раствора сульфата цинка в качестве осадителя мешающих несахаров находят соотношение эквивалентных объемов приготовленного раствора и предназначенного к комплексному использованию 1 моль/дм3 (1 н) раствора NaOH или KOH. Для этого отмеривают пипеткой 10 см3 приготовленного реактива, разбавляют примерно двойным объемом дистиллированной воды, добавляют 3 капли раствора фенолфталеина и оттитровывают 1 моль/дм3 (1 н) раствором NaOH до слабо-розовой окраски. Образующийся в процессе титрования осадок не влияет на титрование.
На этикетках, наклеенных на склянки с растворами, указывают значение полученного соотношения.
Растворы с массовой долей 30 %, 20 % и 10 %. Готовят растворением соответственно 300, 200 или 100 г сульфата цинка 700, 800 или 900 см3 дистиллированной воды.
9.3.2. Бихромат калия (K2Cr2O7), х.ч. или ч.д.а., по ГОСТ 4220-75, 0,017 моль/дм3 (0,1 н) раствор: 4,9033 г перекристаллизованного и высушенного при 150 °C бихромата калия растворяют дистиллированной водой в мерной колбе вместимостью 1000 см3. Перекристаллизацию бихромата калия проводят путем растворения его в кипящей воде до насыщения, затем горячий раствор фильтруют и охлаждают. Выпавшие кристаллы отфильтровывают на стеклянном фильтре, сушат в течение 2 - 3 ч при 100 - 105 °C в сушильном шкафу, измельчают и досушивают при 150 °C в течение 10 - 12 ч до постоянной массы.
Раствор 0,017 моль/дм3 (0,1 н) бихромата калия можно приготовить из фиксанала.
9.3.3. Гексацианоферрат (II) калия (желтая кровяная соль) (K4Fe(CN)6∙3H2O), раствор с массовой долей 15 %: 150 г соли растворяют в 850 см3 воды.
9.3.4. Сульфат меди (II) (медный купорос) (CuSO4∙5H2O), х.ч. или ч.д.а., по ГОСТ 4165-78, раствор с массовой долей 7 %: 70 г сульфата меди (II) растворяют в 930 см3 дистиллированной воды.
9.3.5. Карбонат натрия (безводный) или кристаллический (NaCO3 или Na2CO3∙10H2O), х.ч. или ч.д.а., по ГОСТ 83-79, раствор с массовой долей 15 %: 150 г безводного карбоната натрия или 405 г кристаллогидрата растворяют в 850 или 595 см3 воды.
9.3.6. Йодид калия (KJ), х.ч. или ч.д.а., по ГОСТ 4232-74, растворы с массовой долей 30 %, 20 % и 10 %. Готовят растворением соответственно 300, 200 или 100 г йодида калия в 700, 800 или 900 см3 дистиллированной воды. Хранят в склянке из темного стекла.
9.3.7. Хлорид натрия (поваренная соль) (NaCl), х.ч., по ГОСТ 4233-77, 0,1 моль/дм3 (0,1 н) раствор, удобно готовить из фиксанала.
9.3.8. Хромат калия (K2CrO4), х.ч. или ч.д.а., по ГОСТ 4459-76, раствор с массовой долей 10 % и насыщенный раствор готовят растворением соответственно 10 и 38,6 г хромата калия в 90 и 100 см3 дистиллированной воды.
9.3.9. Нитрат серебра (AgNO3), х.ч. или ч.д.а., по ГОСТ 1277-75, 0,05 или 0,1 моль/дм3 (0,05 или 0,1 н) растворы.
Взвешивают на часовом стекле с точностью до 0,1 г 8,5 или 10,99 г нитрата серебра, переносят в мерную колбу вместимостью 1000 см3, растворяют в воде и доводят объем жидкости при 20 °C до метки.
Поправочный коэффициент устанавливают по 0,1 моль/дм3 (0,1 н) раствору хлорида натрия, приготовленному из фиксанала. Для этого отбирают пипеткой 10 см3 0,1 моль/дм3 (0,1 н) раствора хлорида натрия (NaCl), переносят в коническую колбу, добавляют 2 - 3 капли индикатора - насыщенного раствора хромата калия (п. 9.3.8) и титруют раствором нитрата серебра до перехода цвета жидкости из чисто желтого со взмученным осадком в красновато-бурый.
Эквивалентная концентрация раствора AgNO3 (H1) рассчитывается исходя из формулы:
|
|
(113) |
где:
H - эквивалентная концентрация раствора NaCl;
V - объем раствора NaCl, взятый для титрования, см3;
V1 - объем раствора AgNO3, пошедший на титрование раствора NaCl, см3.
Титр раствора AgNO3 рассчитывают по формуле:
|
|
(114) |
где:
Э - эквивалент AgNO3, равный 169,89 г;
H1 - эквивалентная концентрация раствора AgNO3.
Коэффициент поправки к титру раствора AgNO3 рассчитывают по формуле:
|
|
(115) |
где:
Т - титр раствора AgNO3, г;
Тт - теоретический титр 0,05 моль/дм3 (0,05 н) или 0,1 моль/дм3 (0,1 н) раствора, равный 0,008498 или 0,016989 г.
9.3.10. Ацетат меди (II), х.ч. или ч.д.а., по ГОСТ 5852-79, насыщенный раствор: 8 - 9 г ацетата меди (II) растворяют в 100 см3 дистиллированной воды.
9.4. Основные растворы для определения содержания сахаров
9.4.1. Реактив Фелинга. Состоит из двух растворов. Оба раствора хранят отдельно и смешивают равные их объемы перед употреблением. Разделение и хранение растворов вызвано тем, что двухвалентная медь способна медленно окислять калий-натрий виннокислый в щелочной среде с выделением осадка закиси меди.
Раствор Фелинга 1 (по Бертрану). Исходным реактивом является сульфат меди (II) (CuSO4∙5H2O), х.ч. или ч.д.а., по ГОСТ 4165-76: 40 г перекристаллизованного, не содержащего железа, сульфата меди (II) растворяют в дистиллированной воде в мерной колбе вместимостью 1000 см3. Для перекристаллизации 70 г сульфата меди (II) растворяют, при нагревании, в 100 см3 дистиллированной воды. К раствору добавляют 1 - 2 капли концентрированной азотной кислоты на каждые 100 см3 раствора для окисления возможной примеси железа, доводят до кипения и горячий раствор фильтруют через складчатый фильтр; при этом можно пользоваться воронкой для горячего фильтрования. Фильтрат охлаждают. Для освобождения образовавшихся в фильтрате кристаллов сульфата меди (II) раствор фильтруют через стеклянный фильтр с помощью водоструйного насоса или сливают декантацией. Кристаллы сульфата меди (II) отжимают и высушивают между листами фильтровальной бумаги.
Раствор Фелинга 2 (по Бертрану). Исходными реактивами являются тартрат калия-натрия (сегнетова соль) KNa3C4H4O6∙4H2O, х.ч., по ГОСТ 5845-70 и гидроксид натрия (NaOH), х.ч. или ч.д.а., по ГОСТ 4326-77: 200 тартрата калия-натрия взвешивают с точностью до 0,01 г, растворяют при слабом нагревании в 300 - 400 см3 дистиллированной воды, прибавляют 150 г гидроксида натрия или 200 г гидроксида калия, растворенного в 300 - 350 см3 дистиллированной воды в фарфоровом стакане, и охлаждают. Оба раствора количественно переносят в мерную колбу вместимостью 1000 см3 и доводят дистиллированной водой до метки.
9.4.2. Раствор сульфата аммония железа (III) (железоаммонийные квасцы). Исходным реактивом является сульфат аммония железа (III) NH4Fe(SO4)2∙12H2O, х.ч. или ч.д.а., по ГОСТ 4205-77; 86 г квасцов растворяют в фарфоровом стакане в 600 - 700 см3 воды и осторожно добавляют 200 г (108 см3) концентрированной серной кислоты. После охлаждения раствор переносят в мерную колбу на 1 дм3 доводят до метки водой и перемешивают. Раствор квасцов не должен содержать соли железа (II); при добавлении к 20 см3 раствора 1 - 2 капель перманганата калия розовая окраска должна сохраняться в течение 1 мин. Если окраска исчезает сразу, раствор квасцов окисляют перманганатом калия до появления устойчивой слабо-розовой окраски.
9.4.3. Перманганат калия. 0,02 моль/дм3 (0,1 н) раствор. Исходными реактивами являются перманганат калия KMnO4, х.ч, по ГОСТ 20490-75, оксалат аммония, х.ч., по ГОСТ 5712-78, или оксалат натрия, х.ч. или ч.д.а., по ГОСТ 5839-77, или щавелевая кислота, х.ч. или ч.д.а., по действующей нормативно-технической документации: 3,16 г перманганата калия растворяют в прокипяченной (для удаления CO2 и O2) горячей дистиллированной воде в мерной колбе вместимостью 1000 см3. Раствор охлаждают до 20 °C и доводят водой до метки. Подготовленный таким образом раствор можно употреблять на следующий день. Раствор хранят в склянке из темного стекла.
Для установления титра раствора перманганата калия на аналитических весах на часовом стекле взвешивают 0,1400 г перекристаллизованного оксалата аммония, переносят количественно в коническую колбу и растворяют в 100 см3 воды; добавляют в колбу 2 см3 концентрированной серной кислоты, раствор нагревают до 80 °C на водяной бане и титруют из бюретки раствором перманганата калия при постоянном помешивании до появления розовой окраски, не исчезающей в течение 1 мин. Раствор до окончания титрования должен быть горячим.
Титр раствора перманганата калия по меди в мг вычисляют по формуле:
|
|
(116) |
где:
m - масса навески щавелевокислого аммония, г;
V - количество раствора перманганата калия, пошедшее на титрование, см3;
0,8951 - коэффициент пересчета оксалата аммония на медь;
1000 - перевод в мг.
Примечание. Титр раствора перманганата калия устанавливают также по оксалату натрия (предварительно освобожденному от гигроскопической воды путем нагревания при 120 °C) или по свежеперекристаллизированной щавелевой кислоте с соблюдением тех же условий выполнения, что и при использовании оксалата аммония. При вычислении титра, в случае применения оксалата натрия, следует вместо коэффициента 0,8951 в формулу вводить 0,9488, а в случае применения щавелевой кислоты - 1,0086. Желательно титр устанавливать по двум реактивам (кислоте и аммонию), добиваясь идентичных результатов.
9.4.4. Щелочной медно-цитратный раствор. Исходными реактивами являются сульфат меди (II), х.ч. или ч.д.а., по ГОСТ 4165-78, лимонная кислота (С6H8O7), х.ч., по ГОСТ 3652-69 и карбонат натрия безводный (Na2CO3) или кристаллогидрат (Na2CO3∙10H2O), х.ч. или ч.д.а., по ГОСТ 83-79: 25 г сульфата меди (II) растворяют в 100 см3 дистиллированной воды; 50 г лимонной кислоты растворяют отдельно в 50 см3 дистиллированной воды, 388 г кристаллогидрата карбоната натрия или 143,7 г безводного карбоната натрия также растворяют отдельно в 300 - 500 см3 горячей воды. Раствор лимонной кислоты осторожно вливают небольшими порциями в охлажденный раствор карбоната натрия. После прекращения выделения углекислого газа смесь растворов переносят в мерную колбу вместимостью 1000 см3, вливают в колбу раствор сульфата меди (II), доводят содержимое колбы дистиллированной водой до метки, перемешивают и, если нужно, фильтруют.
9.4.5. Тиосульфат натрия (гипосульфит), 0,1 моль/дм3 (0,1 н) раствор. Исходным реактивом является тиосульфат натрия (Na2S2O3∙5Н2O), х.ч. или ч.д.а., по ГОСТ 4215-66: 25 г тиосульфата натрия взвешивают с точностью до 0,1 г, переносят в мерную колбу вместимостью 1000 см3, растворяют в свежепрокипяченной и охлажденной, без доступа углекислоты, воде. Охлаждают воду в колбе с закрытой пробкой, через которую проходит хлоркальциевая трубка, наполненная натронной известью. В колбу с раствором прибавляют 0,2 г безводного карбоната натрия и доводят объем раствора до метки той же водой. Раствор хранят в склянке из темного стекла.
Коэффициент поправки устанавливают через 8 - 15 дней по 0,017 моль/дм3 (0,1 н) раствору бихромата калия (п. 9.3.2). Раствор можно приготовить из фиксанала, растворяя содержимое ампулы свежепрокипяченной и охлажденной до 20 °C водой. Затем раствор доводят той же водой до метки. В этом случае поправочный коэффициент не устанавливают. Рекомендуется готовить запас раствора тиосульфата натрия в количестве 5 - 10 дм3.
Для установления поправочного коэффициента 0,1 моль/дм3 (0,1 н) раствора тиосульфата натрия в коническую колбу с притертой пробкой или в обычную коническую колбу, закрывающуюся часовым стеклом, из бюретки или пипеткой приливают точно 20 см3 0,017 моль/дм3 (0,1 н) раствора бихромата калия (п. 9.3.2), доливают водой примерно до 100 см3, прибавляют, при помешивании, примерно 4 см3 концентрированной серной колоты и 4 см3 йодида калия, раствор с массовой долей 30 % (п. 9.3.6). Колбу закрывают пробкой или часовым стеклом и оставляют в темном месте на 2 - 3 мин. Затем содержимое колбы титруют раствором тиосульфата натрия, все время интенсивно перемешивая жидкость, пока коричневый цвет раствора не перейдет в светло-желтый. Далее прибавляют 1 см3 раствора крахмала с массовой долей 1 % и продолжают титрование до исчезновения синей окраски и появления зеленоватой окраски соединений трехвалентного хрома. Поправочный коэффициент (K) к точно 0,1 моль/дм3 (0,1 н) раствору находят по формуле:
|
|
(117) |
где:
V - объем 0,1 моль/дм3 (0,1 н) раствора тиосульфата натрия, пошедшего на титрование, см3;
20 - объем точно 0,017 моль/дм3 (0,1 н) раствора бихромата калия, взятого для титрования, см3.
9.4.6. Гексацианоферрат (III) калия (красная кровяная соль), титрованный раствор с массовой долей 1 %. Исходным реактивом является гексацианоферрат (III) калия K3Fe(CN)6, х.ч. или ч.д.а., по ГОСТ 4206-75: 10 г гексацианоферрата (III) калия переносят в мерную колбу вместимостью 1000 см3, растворяют в дистиллированной воде и доводят до метки.
Коэффициент поправки устанавливают следующим образом. В коническую колбу вместимостью 250 см3 с притертой или каучуковой пробкой отбирают пипеткой 50 см3 раствора гексацианоферрата (III) калия с массовой долей 1 %, затем прибавляют 20 см3 раствора сульфата цинка с массовой долей 10 %, не содержащего железа, и 20 см3 раствора йодида калия с массовой долей 20 %, не содержащего свободного йода. Содержимое взбалтывают в закрытой колбе и тотчас же титруют выделившийся йод 0,1 моль/дм3 (0,1 н) раствором тиосульфата натрия в присутствии крахмала в качестве индикатора до его обесцвечивания. Поправку (K) вычисляют по формуле:
|
|
(118) |
где:
V - объем точно 0,1 моль/дм3 (0,1 н) раствора тиосульфата натрия, пошедшего на титрование выделившегося йода, см3;
0,03291 - количество гексацианоферрата (III) калия, соответствующее 1 см3 0,1 моль/дм3 (0,1 н) раствора тиосульфата натрия, г;
0,5 - количество гексацианоферрата (III) калия, содержащегося в 50 см3 точно раствора с массовой долей 1 %.
9.5. Основные растворы для определения содержания витамина С, сернистого ангидрида и достаточности термической обработки
9.5.1. Натриевая соль 2,6-дихлорфенолиндофенола (индикатор), 0,001 моль/дм3 (0,001 н) раствор. Исходными реактивами являются натриевая соль 2,6-дихлорфенолиндофенола, сульфат аммоний железа (II) или соль Мора (NH4)2Fe(SO4)2∙6H2O х.ч., по ГОСТ 4208-72; оксалат натрия, х.ч., по ГОСТ 5839-77 или оксалат аммония, х.ч., по ГОСТ 5712-78: 0,2000 г индикатора растворяют в 700 см3 дистиллированной воды при энергичном взбалтывании и добавляют 300 см3 фосфатного буферного раствора.
Приготовленный раствор фильтруют и хранят в склянке из темного стекла в темном месте в течение 1 - 1,5 месяца.
Поправку к титру устанавливают 1 раз в неделю по 0,01 моль/дм3 (0,01 н) раствору соли Мора. В коническую колбу вместимостью 50 - 100 см3 наливают пипеткой 10 см3 приготовленного раствора индикатора, добавляют 5 см3 насыщенного раствора оксалата натрия (7 г на 100 см3 воды) или аммония и титруют из микробюретки или микропипетки 0,01 моль/дм3 (0,01 н) раствором соли Мора до отчетливого перехода синей окраски в лимонно-желтую (нерезкая перемена окраски указывает на непригодность реактива).
Поправочный коэффициент вычисляют по формуле:
|
|
(119) |
где:
V - количество кубических сантиметров раствора соли Мора, пошедшее на титрование 10 см3 раствора 2,6-дихлорфенолиндофенола;
K1 - поправка к 0,01 моль/дм3 (0,01 н) раствору соли Мора;
0,01 - концентрация раствора соли Мора, моль/дм3;
0,001 - концентрация раствора 2,6-дихлорфенолиндофенола, моль/дм3;
10 - объем раствора 2,6-дихлорфенолиндофенола, см3.
9.5.2. Соль Мора, 0,01 моль/дм3 (0,01 н) раствор.
Исходными реактивами являются соль Мора (NH4)2Fe(SO4)2∙6H2O, х.ч., по ГОСТ 4208-72; перманганат калия (KMnO4), х.ч., по ГОСТ 20490-75 и серная кислота (H2SO4) плотностью 1,84 г/см3, х.ч. или ч.д.а., по ГОСТ 4204-77: 3,92 г соли Мора переносят в мерную колбу вместимостью 1000 см3, растворяют в 0,01 моль/дм3 (0,02 н) раствора серной кислоты (п. 9.1.1). Раствор соли Мора хранят в склянке из темного стекла.
Поправку к титру раствора соли Мора устанавливают по титрованному 0,002 моль/дм3 (0,01 н) раствору марганцовокислого калия через каждые 3 - 4 недели.
В коническую колбу вливают пипеткой 10 см3 приготовленного раствора соли Мора, прибавляют 1,5 см3 серной кислоты, разбавленной в соотношении 1:2, и титруют (не нагревая) 0,002 моль/дм3 (0,01 н) раствором перманганата калия до появления слабо-розового окрашивания.
Поправочный коэффициент вычисляют по формуле:
|
|
(120) |
где:
V1 - количество миллилитров раствора перманганата калия, пошедшее на титрование 10 см3 соли Мора;
10 - объем раствора соли Мора, взятый для титрования, см3;
K2 - поправочный коэффициент к 0,002 моль/дм3 (0,01 н) раствору перманганата калия.
9.5.3. Перманганат калия 0,002 моль/дм3 (0,01 н) раствор. Исходными реактивами являются перманганат калия, х.ч., по ГОСТ 20490-75 или оксалаты аммония или натрия, х.ч., по ГОСТ 5712-78, ГОСТ 5839-77 и серная кислота плотностью 1,84 г/см3, х.ч. или ч.д.а., по ГОСТ 4204-77. Взвешивают на часовом стекле 0,3160 г перманганата калия, растворяют в колбе вместимостью 1000 см3 в горячей свежепрокипяченной дистиллированной воде и доводят, после охлаждения до 20 °C, до метки. 0,002 моль/дм3 (0,01 н) раствор перманганата калия можно приготовить, разбавляя в 10 раз 0,02 моль/дм3 (0,1 н) раствор (п. 9.4.3). Используют коэффициент поправки 0,02 моль/дм3 (0,1 н) раствора.
Поправку к титру 0,002 моль/дм3 (0,01 н) раствора перманганата калия устанавливают по точно 0,005 моль/дм3 (0,01 н) растворам химически чистых оксалатов натрия или аммония и проверяют через 3 - 4 недели.
Для приготовления точно 0,005 моль/дм3 (0,01 н) раствора оксалата натрия или аммония на часовом стекле взвешивают точно 0,0670 г химически чистого, высушенного при 120 °C оксалата натрия или 0,0620 г перекристаллизованного оксалата аммония, переносят в колбу вместимостью 100 см3 бидистиллятом и доводят до метки при 2 °C.
К 10 см3 0,005 моль/дм3 (0,01 н) раствора оксалата натрия или аммония прибавляют 2,5 см3 серной кислоты 1:2. Титрование этих растворов производят при нагревании на водяной бане при 80 - 90 °C, не допуская кипения, до появления слабо-розового окрашивания, не исчезающего в течение 1 мин. Раствор до окончания титрования должен быть горячим.
Поправочный коэффициент вычисляют по формуле:
|
|
(121) |
где V2 - количество кубических сантиметров 0,01 моль/дм3 (0,01 н) раствора перманганата калия, пошедшего на титрование 10 см3 точно 0,005 моль/дм3 (0,01 н) раствора оксалата натрия или аммония.
9.5.4. Приготовление бидистиллята. В колбу вместимостью 2 дм3 заливают дистиллированную воду, прибавляют 0,1 г перманганата калия и несколько капель концентрированной, химически чистой серной кислоты плотностью 1,84 г/см3. Колбу соединяют с помощью каплеуловителя с холодильником и проводят перегонку.
9.5.5. Фосфатный буферный раствор. Исходными реактивами являются дигидрофосфат калия (KH2PO4), х.ч., по ГОСТ 4198-75 и гидрофосфат натрия (Na2HPO4), х.ч., по ГОСТ 4172-76; 0,908 г KH2PO4 растворяют в 100 см3 дистиллированной воды (раствор 1); 2,9687 г Na2HPO4 растворяют в 250 см3 воды (раствор 2). Для приготовления фосфатного буферного раствора соединяют 90 см3 раствора 1 и 210 см3 раствора 2 и хорошо перемешивают.
9.5.6. Раствор йода 0,01 моль/дм3 (0,01 н). Исходными реактивами являются йодид калия, х.ч., по ГОСТ 4232-74; бихромат калия, х.ч., по ГОСТ 4220-75 и концентрированная соляная кислота плотностью 1,19 г/см3, х.ч., по ГОСТ 3118-77. Для практических целей 0,01 моль/дм3 (0,01 н) раствор йода готовят путем соединения заранее приготовленных растворов бихромата калия (1,9600 г в 1000 см3 раствора), йодида калия с массовой долей 10 % и соляной кислоты (плотностью 1,19) в количествах соответственно 25 см3, 2,5 см3 и 5 см3 в мерной колбе на 100 см3, которую затем доливают водой до метки при 20 °C и взбалтывают*(11). Тщательно приготовленный раствор не нуждается в проверке титра.
________________
*(11) Навеску бихромата калия взвешивают на аналитических весах. Раствор бихромата калия, йодида калия и соляной кислоты вносят в колбу пипеткой.
Раствор готовят из расчета дневной потребности в нем. Хранят в посуде из темного стекла с пришлифованной пробкой.
Можно готовить 0,01 моль/дм3 (0,01 н) раствор йода путем разведения в 10 раз 0,1 моль/дм3 (0,1 н) раствора, приготовленного из фиксанала. Для этого отмеривают пипеткой 25 см3 0,1 моль/дм3 (0,1 н) раствора йода, переносят в колбу на 250 см3, доводят объем до метки дистиллированной водой и тщательно перемешивают. Используют сразу же после приготовления.
9.5.7. Ацетатный буферный раствор (pH 4,9). Исходными растворами являются концентрированная уксусная кислота, х.ч., по ГОСТ 61-75 и ацетат натрия безводный или кристаллический, х.ч., по ГОСТ 199-78: 12 г ледяной уксусной кислоты растворяют в колбе вместимостью 1000 см3 и доводят до метки (раствор 1); 16,4 г безводного ацетата натрия, или 27,2 г - с тремя молекулами воды, или 38 г - с шестью молекулами воды растворяют в колбе вместимостью 1000 см3 (раствор 2). Для приготовления ацетатного буферного раствора смешивают растворы 1 и 2 в соотношении 3,5:6,5. Ацетатный буферный раствор стоек и не требует специальных условий хранения.
9.5.8. Пероксид водорода (перекись водорода), раствор с массовой долей 1 %. Исходным реактивом является пероксид водорода с массовой долей 30 - 35 % (пергидроль), х.ч., по ГОСТ 10929-76.
Вначале проверяют фактическую концентрацию имеющегося в наличии пероксида водорода методом, указанным в ГОСТ 10929-76. Затем готовят раствор с массовой долей 1 % путем разбавления определенной навески концентрированного пероксида водорода дистиллированной водой. Например, в результате анализа установлена фактическая концентрация раствора с массовой долей 30 %. Исходя из пропорции, определяем величину навески, необходимую для разбавления:
|
100 - 30 |
|
|
|
Х - 1 |
|
Взвешивают в бюксе 3,4 г пергидроля и разбавляют в 96,6 см3 дистиллированной воды.
Раствор хранят в склянке из темного стекла с пришлифованной пробкой. Готовят по мере надобности, в небольшом количестве.
9.5.9. Бариевая соль паранитрофенилфосфата. Исходными реактивами являются бариевая соль паранитрофенилфосфата, концентрированная соляная кислота плотностью 1,19, х.ч., по ГОСТ 3118-77 и эфир этиловый, х.ч., по действующей нормативно-технической документации. 0,800 г бариевой соли паранитрофенилфосфата растворяют без нагревания в 100 см3 0,001 моль/дм3 (0,001) раствора соляной кислоты*(12).
Нерастворившуюся часть отфильтровывают. Если раствор имеет желтую окраску, то его несколько раз взбалтывают в делительной воронке с равным количеством эфира до обесцвечивания водного слоя. После этого водный слой отделяют от эфира и хранят в склянке из темного стекла в холодильнике.
________________
*(12) 0,001 моль/дм3 (0,001 н) раствор соляной кислоты готовят разбавлением 0,1 моль/дм3 (0,1 н) раствора, приготовленного из фиксанала, в 100 раз дистиллированной водой.
9.5.10. Ацетатный буферный раствор (pH 5,4). Исходными реактивами являются кислота уксусная с массовой долей 80 %, х.ч., по ГОСТ 6968-76 и ацетат натрия, х.ч., по ГОСТ 199-78. 1 моль/дм3 (1 н) раствор уксусной кислоты готовят разбавлением водой 70 см3 концентрированной уксусной кислоты с массовой долей 80 % плотностью 1,07 г/см3 в мерной колбе вместимостью 1000 см3 и доведением до метки. 1 моль/дм3 (1 н) раствор ацетата натрия готовят растворением в воде 136,3 г соли в колбе вместимостью 1000 см3 и доведением до метки. Ацетатный буферный раствор готовят путем смешивания 1 части 1 моль/дм3 (1 н) раствора уксусной кислоты с 5 частями 1 моль/дм3 (1 н) раствора ацетата натрия и проверяют pH среды с помощью pH-метра, или потенциометра, или с помощью универсальной индикаторной бумаги.
9.6. Индикаторы
9.6.1. Бромтимоловый синий, раствор с массовой долей 0,1 %. Интервал перехода окраски 6,0 - 7,6. 0,1 г реактива растворяют в 100 см3 воды или 20-процентного спирта.
9.6.2. Метиловый оранжевый по ГОСТ 10816-64, раствор с массовой долей 1 %. Интервал перехода окраски 3,1 - 4,4. 1 г метилового оранжевого растворяют в 99 см3 дистиллированной воды.
9.6.3. Метиловый красный по ГОСТ 5853-51, раствор с массовой долей 0,2 %. Интервал перехода окраски 4,4 - 6,2. 0,2 г метилового красного растворяют в 60 см3 этилового спирта и доводят водой до 100 см3.
9.6.4. Метиленовый голубой, раствор с массовой долей 1 %. 1 г реактива растворяют в 99 см3 дистиллированной воды.
9.6.5. Тимолфталеин, спиртовой раствор с массовой долей 0,1 %. Интервал перехода окраски 9,4 - 10,6. 0,1 г реактива растворяют в 100 см3 90-процентного этилового спирта.
9.6.6. Фенолфталеин по ГОСТ 5350-72, спиртовой раствор с массовой долей 1 %. Интервал перехода окраски 8,2 - 10,0. 1 г фенолфталеина растворяют в 99 см3 60 - 90-процентного этилового спирта.
9.6.7. Смешанный индикатор. Состоит из двух растворов: метиленового голубого, водного раствора с массовой долей 0,1 % (раствор 1), и метилового красного по ГОСТ 5853-51, раствора с массовой долей 0,02 % (раствор 2).
Раствор 2 готовят растворением 0,02 г метилового красного в 100 см3 60-процентного этилового спирта. К 25 см3 раствора 1 добавляют 3 см3 раствора 2.
9.6.8. Раствор Люголя. Исходными реактивами являются йодид калия, х.ч., по ГОСТ 4232-74 и йод кристаллический, х.ч., по ГОСТ 4159-79. В химическом стакане вместимостью 100 см3 взвешивают 2 г йодида калия, добавляют 15 см3 дистиллированной воды и 1,27 г кристаллического йода. После растворения йода раствор переносят в мерную колбу вместимостью 100 см3 и доводят дистиллированной водой до метки. Раствор хранят в темной склянке с притертой пробкой.
9.6.9. Раствор крахмала с массовой долей 1 %. Исходными реактивами являются крахмал растворимый по ГОСТ 10163-76 и хлорид натрия, х.ч., по ГОСТ 4233-77. 1 г крахмала смешивают с 20 см3 дистиллированной воды и полученный раствор вливают в 80 см3 кипящей воды, помешивая его палочкой. Кипятят 1 мин., затем охлаждают.
Раствор можно приготовить следующим образом: 1 г крахмала смешивают вначале с небольшим количеством (около 20 см3) насыщенного раствора хлорида натрия, затем вливают в доведенный до кипения насыщенный раствор соли (примерно 80 см3) с таким расчетом, чтобы общий объем был равен 100 см3, кипятят около 1 мин. и охлаждают. Такой раствор хранится длительное время без изменения.
9.6.10. Раствор йодидокалиевого крахмала. К 100 см3 охлажденного крахмального клейстера с массовой долей 3 % (приготовление см. выше) добавляют 3 г йодида калия, растворенного в небольшом количестве дистиллированной воды (15 - 20 см3). Хранить раствор рекомендуется в темном, прохладном месте не более 5 - 7 дней.
9.6.11. Приготовление индикаторных бумажек. Белую фильтровальную бумагу смачивают в растворе йодидокалиевого крахмала (п. 9.6.10) и высушивают при комнатной температуре в затемненном месте. Хранить индикаторные бумажки рекомендуется в темном конверте. Индикаторные бумажки, которые приобрели буроватый оттенок, непригодны.
9.6.12. Приготовление реактива, содержащего краску судан III. Исходными реактивами являются краска судан III, спирт этиловый по ГОСТ 5962-67, гидроксид аммония (водный раствор аммиака с массовой долей 25 %), х.ч. или ч.д.а., по ГОСТ 3760-79 и метиленовый голубой. В 70 см3 этилового спирта, нагретого до 60 °C, растворяют 0,2 г краски судан III и 0,05 г метиленового голубого. Приливают 10 см3 раствора аммиака с массовой долей 20 - 25 % и 20 см3 воды. Реактив стоек при хранении.
9.6.13. Приготовление специального реактива, содержащего краску судан III. В 80 см3 этилового спирта растворяют 0,05 г краски судан III. В 18 см3 дистиллированной воды растворяют 0,02 г метиленового голубого. Растворы смешивают и добавляют 2 см3 раствора аммиака с массовой долей 15 %.
Приложение 1
Отбор проб кулинарных и кондитерских полуфабрикатов, подготовка их для анализа и исследуемые физико-химические показатели качества
|
№ п/п |
Продукция |
НТД на продукцию |
Стандарт на отбор проб |
Количество вскрываемых единиц от партии (выборка для осмотра и составления средней пробы, исходного образца или общей пробы) |
Средняя или общая проба (исходный образец) |
Масса пробы для физикохимического анализа |
Подготовка проб для физико-химического анализа |
Нормируемые физико-химические показатели |
ГОСТы на методы испытаний |
Количество продукции для контроля массы |
Допустимые отклонения в массе одной и десяти единиц продукции |
||||
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
||||
|
1. |
Полуфабрикаты мясные: |
|
|
|
|
|
|
|
|
|
|
|
|||
|
полуфабрикат замороженный для жарения |
ТУ 10-0201-36-87 |
10 % от объема партии |
400 - 500 г |
200 - 250 г |
Размораживают, образовавшуюся жидкую фазу добавляют к продукту, дважды измельчают в мясорубке |
Массовая доля (в сыром полуфабрикате): |
|
10 шт. |
±3 % |
||||||
|
- влаги |
|||||||||||||||
|
- соли |
|||||||||||||||
|
- сахаров в пересчете на крахмал |
ГОСТ 10574-73 |
||||||||||||||
|
полуфабрикаты мясные натуральные (крупнокусковые, порционные, мелкокусковые) |
ОСТ 49 208-84 |
ОСТ 49 208-84 |
10 % упаковочных единиц от партии, но не менее трех ящиков |
То же |
То же |
Дважды измельчают в мясорубке |
Свежесть |
|
ГОСТы: |
10 порций из разных мест ящиков |
Порционные полуфабрикаты ±3 %; |
||||
|
мелкокусковые: |
|||||||||||||||
|
- для отдельных кусочков 15 - 25 %; |
|||||||||||||||
|
19496-74 |
- для порций массой: |
||||||||||||||
|
250 г ± 7,5 г |
|||||||||||||||
|
500 г ± 15 г |
|||||||||||||||
|
1000 г ± 10 г |
|||||||||||||||
|
полуфабрикаты мясные рубленые (котлеты: московские, домашние, киевские; бифштекс рубленый) |
ОСТ 49 121-84 |
3 % упаковок от партии менее 10 упаковок, 5 % - при большем количестве упаковок в партии |
|
6 шт. массой 50 г или 4 шт. массой 75 г и более |
Растирают в ступке до получения гомогенной массы |
Массовая доля: |
|
10 шт. |
Котлеты: |
||||||
|
1 шт. - ±5 %; |
|||||||||||||||
|
- влаги |
10 шт. - ±2 % |
||||||||||||||
|
- хлеба (кроме бифштекса) |
Бифштекс: |
||||||||||||||
|
- поваренной соли |
1 шт. - ±2 % |
||||||||||||||
|
|
10 шт. - ±1 % |
||||||||||||||
|
полуфабрикаты мясные рубленые (шницель натуральный рубленый, шницель рубленый, котлеты натуральные рубленые, люля-кебаб) |
ТУ 28-19-84 |
ТУ 28-19-84 |
От партии до 100 мест - 3 емкости (из разных рядов и ярусов), от партии свыше 100 мест - на каждые следующие 50 мест дополнительно по одной емкости |
Не менее 600 г |
6 шт. массой 50 г или 4 шт. массой 75 г и более |
Растирают в ступке до получения гомогенной массы |
Массовая доля: |
|
10 шт. |
±2 % для одного полуфабриката. Отклонения в массе 10 шт. полуфабрикатов не допускаются |
|||||
|
- сухих веществ |
|||||||||||||||
|
- поваренной соли |
|||||||||||||||
|
- хлеба с учетом панировочных сухарей (для шницеля рубленого) |
|
||||||||||||||
|
полуфабрикаты из рубленого мяса (котлеты, биточки, шницели, зразы, бифштексы, тефтели) |
Рецептуры сборников |
3 % упаковок от партии менее 10 упаковок, 5 % - при большем количестве упаковок в партии |
10 шт. |
То же |
То же |
То же |
|
То же |
То же |
То же |
|||||
|
фарш мясной (натуральный, особый) фарш мясной |
ТУ 10-02.01.124-90 |
То же |
10 % от объема партии, но не менее 3 ящиков |
То же |
|
То же |
Массовая доля: |
|
|
2 % упаковочных единиц, но не менее 10 партий |
Масса 1 порции фарша, г: |
||||
|
- жира |
250 ± 5; |
||||||||||||||
|
- влаги |
500 ± 10; |
||||||||||||||
|
Рецептуры сборников |
|
То же |
|
500 г |
То же |
|
|
|
20 шт. |
1000 ± 10 |
|||||
|
фрикадельки (приготовляемые на производстве) |
Рецептуры сборников |
- |
Не более 2 % лотков, но не менее одного лотка |
Не менее 1000 г |
650 - 700 г (20 шт.) |
То же |
|
|
|
20 шт. |
|
||||
|
2. |
Кость |
10 упаковочных единиц (ящиков, мешков) |
Не менее 10 кг |
|
|
Массовая доля мякотных тканей. |
|
Для фасованной суповой кости из каждой упаковочной единицы выборки отбирают по одной порции |
|
||||||
|
Посторонние примеси |
|||||||||||||||
|
3. |
Мясные кулинарные изделия: |
|
|
|
|
|
|
|
|
|
|
|
|||
|
мясо шпигованное тушеное (крупным куском для магазинов кулинарии или нарезанное на порции для вторых блюд, в желе) |
ТУ 28-20-84 |
От партии до 100 мест - не менее 3 емкостей, от партии свыше 100 мест - на каждые следующие 50 мест дополнительно по 1 емкости |
Для изделий массой: более 2 кг - две единицы продукции для всех видов испытаний; менее 2 кг - по две единицы для каждого вида испытаний |
400 - 500 г, состоящих из двух точечных проб от разных единиц продукции |
Гомогенизируют в размельчителе тканей или дважды измельчают в мясорубке, перемешивают до получения однородной массы |
Массовая доля: |
|
ГОСТ 9957-73 Взвешиванием |
- |
- |
|||||
|
- поваренной соли |
|||||||||||||||
|
- моркови к массе готового кулинарного изделия |
|||||||||||||||
|
мясо жареное (говядина и свинина жареные крупным куском для холодных блюд и магазинов кулинарии и нарезанные на порции для вторых блюд, в желе) |
ТУ 28-21-84 |
То же |
То же |
То же |
То же |
То же |
Массовая доля поваренной соли |
|
- |
- |
|||||
|
говядина отварная |
ТУ 28-22-84 |
То же |
То же |
То же |
То же |
То же |
То же |
|
То же |
- |
- |
||||
|
4. |
Полуфабрикаты из мяса птицы: |
|
|
|
|
|
|
|
|
|
|
|
|||
|
полуфабрикаты из мяса кур (тушка, филе, окорочок, набор для бульона) |
ГОСТ 49138-85 |
5 % от количества транспортных упаковок в партии, но не менее одной |
3 единицы продукции, а для набора - части трех или более тушек |
|
То же |
Свежесть |
|
|
2 % от выборки, но не менее 10 упаковок |
±4 г для единицы продукции в потребительской таре |
|||||
|
Температура |
|||||||||||||||
|
ОСТ 49-138-85 |
|||||||||||||||
|
Масса нетто единицы продукции в потребительской таре |
|
||||||||||||||
|
полуфабрикаты из мяса уток и утят (окорочок, тушка, грудка, набор утиный, кожа шеи для фарширования) |
ОСТ 1002-01-0587 |
ОСТ 10-0201-05-87 |
5 % от количества транспортных упаковок в партии, но не менее одной |
3 единицы продукции для фасованных полуфабрикатов, для весовых - не менее трех |
|
Гомогенизируют в размельчителе тканей или дважды измельчают в мясорубке, перемешивают до получения однородной массы |
То же |
|
2 % выборки, но не менее трех упаковочных единиц |
±4 г для порции полуфабриката в полимерной пленке, ±10 г для порции полуфабриката, упакованного в лоток из полимерных материалов |
|||||
|
ОСТ 10-02-0105-87 |
|||||||||||||||
|
полуфабрикаты из мяса птицы. Цыплята любительские, цыплята-табака |
ОСТ 49 131-85 |
То же |
То же |
|
То же |
Свежесть |
|
2 % упаковок от выборки, но не менее 10 |
±4 г от массы, указанной на этикетке |
||||||
|
Массовая доля поваренной соли |
|||||||||||||||
|
Масса единицы продукции |
ОСТ 49-131-85 |
||||||||||||||
|
Температура в толще грудной мышцы |
|
||||||||||||||
|
котлеты особые из птицы |
ТУ 28-49-90 |
ТУ 28-49-90 |
От партии до 100 мест - 3 единицы упаковки, от партии свыше 100 мест - на каждые следующие 50 мест дополнительно по 1 единице упаковки |
|
3 шт. массой 100 г или 6 шт. массой 50 г |
Вместе с панировочными сухарями растирают в ступке, или дважды измельчают в мясорубке, или гомогенизируют в аппарате для измельчения тканей, перемешивают |
Массовая доля: |
или |
10 шт. |
Для 1 шт.: |
|||||
|
- влаги |
ГОСТ 8756.2-82 |
±2,5 г |
|||||||||||||
|
- хлеба с учетом панировочных сухарей |
±3 % - при формовке вручную 10 шт. не должны иметь отклонений в меньшую сторону |
||||||||||||||
|
- жира |
|
||||||||||||||
|
- поваренной соли |
|
||||||||||||||
|
5. |
Кулинарные изделия из птицы: |
|
|
|
|
|
|
|
|
|
|
|
|||
|
птица отварная |
ТУ 28-23-84 |
От партии до 100 мест - 3 единицы упаковки от партии свыше 100 мест - на каждые следующие 50 мест дополнительно по 1 единице упаковки |
От птиц массой: более 2 кг - две единицы продукции для всех видов испытаний. Менее 2 кг - по две единицы для каждого вида испытаний |
400 - 500 г, состоящих из двух точечных проб от разных единиц продукции |
Гомогенизируют в аппарате для измельчения тканей или дважды измельчают в мясорубке, перемешивают |
Массовая доля поваренной соли |
|
|
|
||||||
|
мякоть птицы отварная в форме брикета |
ТУ 28-34-84 |
То же |
То же |
То же |
То же |
То же |
То же |
|
То же |
|
|
||||
|
6. |
Полуфабрикаты рыбные: |
|
|
|
|
|
|
|
|
|
|
|
|||
|
полуфабрикаты специальной разделки незамороженные, мороженые, филе рыбное мороженое |
ОСТ 15-37-72 |
Не менее трех в соответствии с таблицей |
Для составления общей пробы из разных мест каждой вскрытой ед. упаковки берут по три разовых пробы массой до 0,5 кг - общей массой около 1,5 кг. Разовые пробы соединяют. При расфасовке в коробки, пакеты отбирают 1 - 2 расфасовки от каждой ед. упаковки и взвешивают поштучно |
Отбирают из общей пробы при массе экземпляра до 100 г - не более 0,5 кг, до 1 кг - 3 рыбы более 1 кг - из трех рыб вырезают по 3 поперечных куска мяса общей массой 0,5 кг. Три единицы расфасовки |
Мелкую рыбу дважды измельчают в мясорубке целиком, а крупную - после удаления кожи и костей |
То же |
|
|
|
||||||
|
масса нетто продукта в ед. упаковки, кг |
к-во отбираемых ед. упак., % от партии |
||||||||||||||
|
до 25 |
1,0 |
||||||||||||||
|
свыше 25 до 50 |
2,0 |
||||||||||||||
|
свыше 50 до 100 |
5,0 |
||||||||||||||
|
свыше 100 - 150 |
7,0 |
||||||||||||||
|
фарш мороженый |
ОСТ 1581-71 |
То же |
То же |
Из разных мест среднего в ящике блока мороженого фарша отбирают 0,4 - 0,5 кг |
Пропускают через мясорубку |
|
|
|
|
|
|||||
|
полуфабрикаты специальной разделки мороженые, расфасованные в потребительскую упаковку (коробки, пакеты) |
То же |
То же |
1 - 2 единицы потребительской упаковки от каждой вскрытой единицы |
3 единицы потребительской упаковки |
|
|
|
|
|
|
|||||
|
ГОСТ 3948-69 |
|
|
|
|
|
|
|||||||||
|
биточки рыбные |
ТУ 28-40-84 |
От партии до 100 мест - 3 единицы упаковки, от партии свыше 100 мест на каждые следующие 50 мест дополнительно по 1 единицы упаковки |
10 шт. |
10 шт. |
Растирают в ступке до получения гомогенной массы |
Массовая доля: |
|
|
10 шт. |
Масса 10 шт. полуфабрикатов не должна иметь отклонений |
|||||
|
- хлеба |
|
|
|||||||||||||
|
- поваренной соли |
|
|
|||||||||||||
|
7. |
Кулинарные изделия из рыбы: |
|
|
|
|
|
|
|
|
|
|
|
|||
|
тефтели, фрикадельки рыбные |
ТУ 28-39-84 |
То же |
13 шт. |
10 шт. |
То же |
То же и кислотность в пересчете на яблочную кислоту |
|
То же и ГОСТ 8756.15-70 |
То же |
Масса 10 шт. изделий не должна иметь отклонений |
|||||
|
рыба отварная семейства осетровых |
ТУ 28-27-84 |
Не менее 10 % всего количества упаковок от партии |
3 куска рыбы или 3 рыбы общей массой не более 0,6 кг |
|
Массовая доля поваренной соли рН рыбы |
|
2 % от общего количества упаковок в партии, но не менее 10 упаковок выборки |
|
|||||||
|
ГОСТ 8756.15-70 |
|||||||||||||||
|
Посторонние примеси |
|
Визуально |
|||||||||||||
|
8. |
Бульоны: |
|
|
|
|
|
|
|
|
|
|
|
|||
|
бульоны пищевые |
ОСТ 49 201-83 |
ОСТ 49 201-83 |
3 % от объема партии, но не менее 2 упаковочных единиц |
Для жидких бульонов - не менее 500 см3, для сухих бульонов - не менее 1000 г |
500 см3 жидкого бульона, 500 г сухого бульона |
Жидкий и концентрированный бульоны нагревают до 60 °С и тщательно перемешивают |
Массовая доля: |
|
|
- |
- |
||||
|
- сухих веществ |
|
ГОСТ 8756.2-82 |
|
|
|||||||||||
|
- поваренной соли |
|
ГОСТ 8756.20-70 |
|
|
|||||||||||
|
- жира |
|
|
|
||||||||||||
|
бульоны костные концентрированные |
ТУ 28-18-84 |
ТУ 28-18-84 |
От партии до 100 мест - не менее 3 емкостей, от партии свыше 100 мест - на каждые мест дополн емкости |
Не менее 600 г |
300 г |
Нагревают до 65 - 70 °С. Тщательно перемешивают |
Массовая доля: |
|
|
- |
- |
||||
|
- сухих веществ |
|
ГОСТ 8756.2-82 |
|
|
|||||||||||
|
- жира |
|
|
|
||||||||||||
|
бульон куриный костный |
ТУ 28-24-84 |
ТУ 28-24-84 |
То же |
То же |
То же |
То же |
Массовая доля сухих веществ |
|
ГОСТ 8756.2-82 |
- |
- |
||||
|
Посторонние примеси |
Визуально |
||||||||||||||
|
бульоны с желатином |
ТУ 28-25-84 |
ТУ 28-25-84 |
То же |
То же |
То же |
То же |
Массовая доля: |
|
|
- |
- |
||||
|
- сухих веществ |
ГОСТ 8756.2-82 |
||||||||||||||
|
- поваренной соли |
ГОСТ 8756.20-82 |
||||||||||||||
|
бульоны |
Рецептуры сборников |
По аналогии с ТУ 28-18 (24, 25)-84 |
То же |
То же |
500 мл |
То же |
То же |
|
То же |
- |
- |
||||
|
9. |
Соусы: |
|
|
|
|
|
|
|
|
|
|
|
|||
|
соусы концентрированные |
ТУ 28-8-84 |
ТУ 28-8-84 |
То же |
То же |
300 г |
|
Массовая доля: |
|
|
- |
- |
||||
|
- сухих веществ |
|||||||||||||||
|
- жира |
|||||||||||||||
|
Общая кислотность |
|||||||||||||||
|
10. |
Овощные полуфабрикаты: |
|
|
|
|
|
|
|
|
|
|
|
|||
|
картофель сырой очищенный |
ТУ 28-47-90 |
ТУ 28-47-90 |
От партии до 10 мест - не менее 1 единицы упаковки, свыше 10 до 20 мест - не менее 3 единиц, от 20 до 50 мест - не менее 5 единиц, от партии свыше 50 мест дополнительно по 1 единице упаковки |
Не менее 3 кг |
10 клубней сульфитированного картофеля и 10 клубней несульфитированного |
Клубни разрезают по двум перпендикулярным осям на четыре части и каждую четвертую часть натирают на мелкой терке, тщательно перемешивают и растирают в ступке до получения однородной массы |
Массовая доля сернистого ангидрида |
ТУ 28-47-90 |
Массу упакованного полуфабриката определяют путем одновременного взвешивания 3 единиц упаковки |
Отклонений массы не допускается |
|||||
|
капуста белокочанная свежая зачищенная, морковь, свекла, лук репчатый свежие очищенные, целые и нарезанные |
ТУ 28-48-90 |
ТУ 28-48-90 |
То же |
Не менее 3 кг корнеплодов и лука, не менее 4 - 5 кочанов капусты, для нарезанных - не менее 600 г |
- |
- |
Масса зачищенных кочанов |
Взвешиванием |
- |
- |
|||||
|
Размер корнеплодов (луковиц) по наибольшему поперечному диаметру |
Измерением |
||||||||||||||
|
Содержание корнеплодов (луковиц) с отклонениями от установленного размера |
Взвешиванием |
||||||||||||||
|
овощи обработанные, нарезанные (редис, редька) |
ТУ 28-13-84 |
ТУ 28-13-84 |
От партии до 100 мест - не менее 3 емкостей, от партии свыше 100 мест - на каждые следующие 50 мест дополнительно по 1 емкости |
Не менее 600 г |
- |
- |
- |
- |
- |
- |
|||||
|
огурцы соленые нарезанные припущенные. Капуста белокочанная свежая нарезанная бланшированная |
ТУ 28-16-84 |
ТУ 28-16-84 |
То же |
То же |
- |
- |
- |
- |
- |
- |
|||||
|
овощи пассерованные |
ТУ 28-28-84 |
ТУ 28-28-84 |
От партии до 100 мест - не менее 3 емкостей, от партии свыше 100 мест – на каждые следующие 50 мест дополнительно по 1 емкости |
Не менее 600 г |
|
Измельчают в мясорубке или в размельчителе тканей, перемешивают |
Массовая доля: |
|
- |
- |
|||||
|
- сухих веществ |
ГОСТ 8756.2-82 |
||||||||||||||
|
- жира |
|||||||||||||||
|
Титруемая кислотность |
ГОСТ 8756.15-70 |
||||||||||||||
|
капуста квашеная тушеная |
ТУ 28-30-84 |
ТУ 28-30-84 |
То же |
Не менее 600 г |
|
То же |
То же |
То же |
- |
- |
|||||
|
свекла маринованная, свекла тушеная для борща |
ТУ 28-10-84 |
ТУ 28-10-84 |
То же |
То же |
|
То же |
Массовая доля: |
|
- |
- |
|||||
|
- сухих веществ |
|||||||||||||||
|
- жира |
|||||||||||||||
|
Общая кислотность |
|||||||||||||||
|
картофель, морковь и свекла отварные (очищенные целые или нарезанные кубиками) |
ТУ 28-9-84 |
ТУ 28-9-84 |
То же |
То же |
- |
- |
- |
- |
- |
- |
|||||
|
полуфабрикаты сбивные из овощей (из свеклы, моркови, тыквы) |
ТУ 28 УССР 266-87 |
То же |
То же |
Не менее 600 г |
Тщательно перемешивают |
Массовая доля: |
|
- |
- |
||||||
|
- влаги |
ГОСТ 8756.2-82 |
||||||||||||||
|
- сахара |
цианидным (объемным) методом |
||||||||||||||
|
запеканки из овощей (капустная, морковная, картофельная с мясом, овощная) |
ТУ 28-37-84 |
ТУ 28-37-84 |
То же |
То же |
|
Гомогенизируют в аппарате для измельчения тканей или дважды измельчают на мясорубке, перемешивают |
Массовая доля: |
|
- |
- |
|||||
|
- сухих веществ |
ГОСТ 8756.2-82 |
||||||||||||||
|
- жира |
|||||||||||||||
|
Посторонние примеси |
Визуально |
||||||||||||||
|
биточки (котлеты) овощные (капустные, морковные, свекольные, картофельные) |
ТУ 28-12-84 |
ТУ 28-12-84 |
То же |
10 шт. |
4 шт. |
То же |
То же |
То же и ТУ 28-12-84 |
10 шт |
Для 1 шт. - ±3 %. Масса 10 шт. полуфабрикатов не должна иметь отклонений |
|||||
|
голубцы, кабачки с мясным фаршем |
Рецептуры сборников |
По аналогии с ГОСТ 4288-76 |
То же |
10 шт. |
4 шт. |
Овощи гомогенизируют в аппарате для измельчения тканей или дважды измельчают на мясорубке. Фарш растирают в ступке до получения однородной массы |
То же и массовая доля: |
|
|
|
|||||
|
- фарша к массе полуфабриката |
То же и взвешиванием |
||||||||||||||
|
- поваренной соли |
ГОСТ 8756.20-70 |
||||||||||||||
|
11. |
Овощные кулинарные изделия: |
|
|
|
|
|
|
|
|
|
|
||||
|
салат из капусты квашеной |
ТУ 28-15-84 |
ТУ 28-15-84 |
От партии до 100 мест - 3 емкости, от партии свыше 100 мест - на каждые следующие 50 мест дополнительно по 1 емкости |
Не менее 600 г |
|
- |
- |
- |
- |
- |
|||||
|
голубцы. Полуфабрикаты и кулинарные изделия |
ТУ 28-38-84 |
То же |
|
|
|
Соотношение капусты (вареной) и фарша |
ТУ 28-38-83 |
10 шт. |
Масса 10 шт. изделий не должна иметь отклонений |
||||||
|
Общая кислотность |
ГОСТ 8756.15-70 |
||||||||||||||
|
Массовая доля: |
|
||||||||||||||
|
- поваренной соли |
ГОСТ 8756.20-70 |
||||||||||||||
|
- жира |
|||||||||||||||
|
- сухих веществ |
ГОСТ 8756.2-82 |
||||||||||||||
|
12. |
Салаты. Полуфабрикаты (мясной, столичный, рыбный) |
ТУ 28-14-84 |
ТУ 28-14-84 |
То же |
То же |
- |
- |
- |
- |
- |
- |
||||
|
13. |
Полуфабрикаты творожные (тесто для сырников 11 видов, тесто для вареников ленивых 5 видов, вареники "Московские" с творогом замороженные 6 видов, вареники "Крестьянские" 2 видов, блинчики крестьянские 4 видов, блинчики с нежирным творогом) |
ОСТ 1002-02-687 |
10 % едиинц транспортной тары с продукцией. При наличии в партии менее 10 единиц отбирают одну. Из каждой единицы транспортной тары отбирают 1 единицу потребительской тары |
Около 500 г |
Около 100 г |
Творожные начинки растирают в ступке до получения однородной консистенции. Тесто тщательно вымешивают. Оболочки блинчиков измельчают ножом, а затем гомогенизируют в размельчителе тканей без добавления воды |
Массовая доля: |
|
|
|
|
||||
|
- влаги |
|
||||||||||||||
|
- жира |
|
||||||||||||||
|
- сахарозы |
|
||||||||||||||
|
- соли |
|
||||||||||||||
|
Кислотность |
|
ГОСТ 3624-67 |
|||||||||||||
|
Температура |
|
|
|||||||||||||
|
Масса продуктов |
|
||||||||||||||
|
14. |
Творожные кулинарные изделия: |
|
|
|
|
|
|
|
|
|
|
||||
|
запеканка и пудинг из творога |
ТУ 28-94-84 |
ТУ 28-94-84 |
От партии до 100 мест - 3 емкости, от партии свыше 100 мест - на каждые следующие 50 мест дополнительно по 1 емкости |
Не менее 600 г |
|
Измельчают на мясорубке или в размельчителе тканей |
Массовая доля: |
|
- |
- |
|||||
|
- сухих веществ |
|||||||||||||||
|
- поваренной соли |
|||||||||||||||
|
- жира |
|||||||||||||||
|
- сахара |
|||||||||||||||
|
Общая кислотность |
ГОСТ 3624-67 |
||||||||||||||
|
15. |
Крупяные полуфабрикаты: |
|
|
|
|
|
|
|
|
|
|
||||
|
биточки (котлеты) крупяные |
ТУ 28-31-84 |
ТУ 28-31-84 |
От партии до 100 мест - 3 единицы упаковки, от партии свыше 100 мест - на каждые следующие 50 мест - дополнительно по 1 единицы упаковки |
10 шт. |
4 шт. |
Гомогенизируют в аппарате для измельчения тканей или дважды измельчают в мясорубке и перемешивают для получения однородной массы |
Массовая доля: |
|
10 шт. |
1 шт. ±3 %. Масса 10 шт. полуфабрикатов не должна иметь отклоненеий |
|||||
|
- сухих веществ |
|||||||||||||||
|
- сахара |
|||||||||||||||
|
Посторонние примеси |
Визуально |
||||||||||||||
|
16. |
Крупяные кулинарные изделия: |
|
|
|
|
|
|
|
|
|
|
||||
|
запеканки крупяные |
ТУ 28-33-84 |
ТУ 28-33-84 |
То же |
Не менее 600 г |
То же |
Массовая доля: |
|
- |
- |
||||||
|
- сухих веществ |
|||||||||||||||
|
- жира (в пересчете на сухое вещество) |
|||||||||||||||
|
17. |
Мучные полуфабрикаты: |
|
|
|
|
|
|
|
|
|
|
||||
|
тесто охлажденное |
ТУ 28-50-90 |
ТУ 28-50-90 |
От партии до 20 мест - единицы упаковки, от 20 до 50 мест - 5 единиц упаковки, свыше 50 мест - на каждые следующие 50 мест дополнительно по 1 единице упаковки |
Не менее 1,5 кг |
Не менее 300 г для слоеного пресного и 700 г для других видов теста |
Лабораторный образец слоеного теста только вымешивают. Лабораторные образцы других видов теста раскатывают в пласт прямоугольной формы, делят по диагоналям, отбирают противоположно лежащие треугольники, соединяют и вымешивают |
Щелочность |
2 % от общего количества фасовок в партии, но не менее 10 единиц |
1 фасовка ± 0,5 %. Отклонение массы 10 фасовок в меньшую сторону не допускается |
||||||
|
Кислотность |
ГОСТ 5670-51 |
||||||||||||||
|
Массовая доля (в пересчете на сухое вещество): |
|
||||||||||||||
|
- влаги |
ТУ 28-91 |
||||||||||||||
|
- сахара |
|||||||||||||||
|
- жира |
|||||||||||||||
|
тесто для оладий, блинов и блинчиков |
Рецептуры сборников |
|
|
Из разных мест кастрюли отбирают 5 порций массой около 40 г |
200 г |
|
|
|
|
|
|
||||
|
пельмени замороженные |
ОСТ 49 120-78 |
ОСТ 49 120-78 |
Не более 1 % упаковок или ящиков, но не менее 3 упаковок или ящиков |
|
Не менее 400 г |
Фарш тщательно перемешивают и измельчают на мясорубке с диаметром отверстий решетки 2 - 3 мм дважды |
Массовая доля: |
|
|
А. По 3 пачки от каждой вскрытой при выборке групповой упаковки |
|
||||
|
- поваренной соли |
|
ГОСТ 9957-73 Взвешиванием |
|||||||||||||
|
- фарша к массе пельменя |
|
ОСТ 49-120-78 |
|||||||||||||
|
- жира в фарше |
|
ОСТ 49-120-78 |
|||||||||||||
|
Толщина тестовой оболочки |
|||||||||||||||
|
Толщина в местах заделки |
|
|
|||||||||||||
|
пельмени (вырабатываемые на производстве) |
Рецептуры сборников |
|
Не менее 3 лотков |
Не менее 1000 г |
650 - 700 г |
То же |
То же |
То же |
- |
- |
|||||
|
блинчики с фаршем |
ТУ 28-17-84 |
ТУ 28-17-84 |
От партии до 100 мест - 3 емкости, от партии свыше 100 мест - на каждые следующие 50 мест дополнительно по 1 емкости |
10 шт. |
5 шт. |
Блинчиковые оболочки измельчают на аппарате для измельчения тканей или на мясорубке, фарш растирают в ступке до получения однородной массы |
Массовая доля: |
|
10 шт. |
1 шт. ±3 % |
|||||
|
- сухих веществ в фарше |
ГОСТ 8756.2-82 |
||||||||||||||
|
- фарша к общей массе блинчиков |
Взвешиванием |
||||||||||||||
|
Полуфабрикаты кондитерские выпеченные: |
|
|
|
|
|
|
|
|
|||||||
|
для штучных пирожных |
Рецептуры сборников |
От партии до 1000 шт. - 8 шт.; от партии свыше 1000 шт. - 13 шт. |
Не менее 200 г |
Не менее 100 г |
|
Измельчают в фарфоровой ступке, на терке, ланцетом или механическим размельчителем (в зависимости от консистенции и структуры продукта) |
|
|
- |
- |
|||||
|
для нарезных пирожных (пласты) |
То же |
То же |
От партии до500 шт. - 2 шт.; от партии свыше 500 шт. - 5 шт. |
1 шт. изделия разрезают под прямым углом на две или четыре равные части и отбирают одну полученную часть |
То же |
|
|
- |
- |
||||||
|
для тортов штучных |
То же |
То же |
То же |
Не менее 1 шт. изделия |
То же |
- |
|
|
- |
- |
|||||
|
для тортов, реализуемых по массе (пласты) |
Рецептуры сборников |
От партии до500 шт. - 2 шт.; от партии свыше 500 шт. - 5 шт. |
1 шт. изделия разрезают под прямым углом на две или четыре равные части и отбирают одну полученную часть |
Не менее 100 г |
То же |
|
|
- |
- |
||||||
|
18. |
Отделочные полуфабрикаты: |
|
|
|
|
|
|
|
|
|
|
||||
|
сироп для промочки изделий |
То же |
То же |
Не менее 500 г |
Не менее 200 г |
Не менее 100 г |
Тщательно перемешивают |
|
|
- |
- |
|||||
|
кремы, помада и др. |
То же |
То же |
То же |
То же |
То же |
То же |
|
|
- |
- |
|||||
|
19. |
Мучные кулинарные изделия: |
|
|
|
|
|
|
|
|
|
|
||||
|
пирожки жареные из дрожжевого теста, вырабатываемые на автоматах |
ТУ 28-11-84 |
ТУ 28-11-84 |
|
Не более 0,4 % от общего количества в партии, но не менее 20 шт. |
5 шт. |
По ¼ основы от каждого из 5 шт. пирожков соединяют, тщательно измельчают острым ножом или на мясорубке с мелкой решеткой. Начинку пирожков соединяют и растирают в ступке до получения однородной массы |
Массовая доля |
|
20 шт. |
Отклонения от установленной массы 20 пирожков не должны превышать: 1,5 % в меньшую сторону, 3 % в большую сторону. Для одного пирожка: 5 % в меньшую сторону, в большую сторону не регламентируется |
|||||
|
- начинки к массе пирожка |
Взвешиванием |
||||||||||||||
|
- влаги в основе |
|||||||||||||||
|
- сухих веществ в начинке |
|||||||||||||||
|
- сахара в пересчете на сухое вещество |
|||||||||||||||
|
- жира в пересчете на сухое вещество в основе |
|||||||||||||||
|
Кислотность основы |
ГОСТ 5670-51 |
||||||||||||||
|
пирожки жареные из дрожжевого теста, вырабатываемые вручную |
РСТ УССР 1765-82 |
РСТ УССР 1765-82 |
- |
0,3 % от всей партии, но не менее 10 шт. |
5 шт. |
То же |
То же |
То же |
10 шт. |
10 шт. ± 3 %, для 1 пирожка не более 5 % в меньшую сторону |
|||||
|
пирожки печеные и жареные из дрожжевого теста ("Столовые" и сдобные) |
Рецептуры сборников |
По аналогии с ГОСТ 5667-65 |
2 - 3 лотка |
0,3 % от всей партии, но не менее 10 шт. |
4 шт. |
Начинку растирают в ступке. Основу измельчают ножом или техническим измельчителем, перемешивают |
То же |
То же |
10 шт. |
|
|||||
|
беляши, чебуреки |
То же |
То же |
То же |
То же |
|
|
То же, кроме кислотности основы (для чебуреков) |
То же, кроме ГОСТ 5670-51 РСТ Каз ССР 917-81 |
10 шт. |
|
|||||
|
Массовая доля лука |
|
5 шт. |
|
||||||||||||
|
кулинарные изделия из рыбы, мяса, запеченные в тесте, штучные |
То же |
То же |
То же |
То же |
То же |
То же |
То же, что и для пирожков жареных из дрожжевого теста |
|
|
|
|||||
|
пироги полуоткрытые штучные и весовые |
То же |
То же |
То же |
От весовых и штучных массой 1 кг - 5 шт.; от штучных массой 0,5 кг - 10 шт. |
Весовых - 1/2 шт.; штучных при массе 1 кг - 1 шт.; массой 0,5 кг - 2 шт. |
То же |
То же |
|
|
|
|||||
|
кулебяки из дрожжевого теста |
То же |
То же |
То же |
То же |
То же |
То же |
То же |
|
|
|
|||||
|
хворост и другие изделия, жаренные во фритюре |
То же |
То же |
То же |
0,3 % всей партии |
|
Измельчают ножом или механическим измельчителем, перемешивают |
Массовая доля влаги |
|
|
||||||
|
Кислотность |
ГОСТ 5670-51 |
|
|
||||||||||||
|
Качество фритюра |
Методы определения качества фритюрного жира |
|
|
||||||||||||
|
20. |
Булочные изделия (сдобные): |
|
|
|
|
|
|
|
|
|
|
||||
|
булочки, слойки с марципаном и др. |
Рецептуры сборников |
2 - 3 лотка |
Из каждых 10 лотков или ящиков при массе изделия менее 1 кг отбирают 0,3 % всей партии, но не менее 10 шт. |
Весовых и штучных: при массе более 400 г - 1 шт.; штучных при массе от 200 до 400 г - не менее 2 шт.; при массе от 100 до 200 г - не менее 3 шт.; менее 100 г - 6 шт. |
Начинку растирают в ступке. Основу (после удаления корочек) измельчают ножом, теркой или механическим измельчителем, перемешивают |
Массовая доля: |
|
|
|
||||||
|
|
|
|
|
|
|
|
- начинки |
Взвешиванием |
|
|
|||||
|
|
|
|
|
|
|
|
- сухих веществ (в начинке) |
Высушивание в сушильном шкафу или в приборе Чижовой |
|
|
|||||
|
|
|
|
|
|
|
|
- влаги (в основе) |
|
|
||||||
|
|
|
|
|
|
|
|
- жира (в основе) |
|
|
||||||
|
|
|
|
|
|
|
|
- сахара (в основе) |
ГОСТ 5672-72 |
|
|
|||||
|
вареники сдобные с творогом |
ГОСТ 24557-81 |
2 - 3 лотка |
Из каждых 10 лотков или ящиков при массе изделия менее 1 кг отбирают 0,3 % всей партии, но не менее 10 шт. |
3 шт. |
То же |
То же |
То же |
|
|
||||||
|
ватрушки сдобные с творогом |
Рецептуры сборников |
То же |
То же |
То же |
4 - 6 шт. |
То же |
То же |
То же |
|
|
|||||
|
сочник с творогом |
То же |
То же |
То же |
То же |
То же |
То же |
То же и щелочность |
ГОСТ 5898-74, |
|
|
|||||
|
ГОСТ 5903-77, |
|||||||||||||||
|
21. |
Мучные кондитерские изделия: |
|
|
|
|
|
|
|
|
|
|
|
|||
|
торты и пирожные |
ТУ 10.04.08.13-88 |
От партии до 50 единиц транспортной тары - 3 единицы; от 51 до 150 единиц - 5 единиц; от 151 до 500 единиц - 8 единиц; от 501 до 1200 единиц - 13 единиц |
Не менее 1 шт. для тортов; не менее 1 шт. изделия в ассортименте каждой единицы транспортной тары для пирожных |
Не менее 100 г |
Массовая доля: |
|
|
Отклонение массы по верхнему пределу не ограничивается. |
|||||||
|
- влаги |
По нижнему пределу: |
||||||||||||||
|
- общего сахара в пересчете на сухое вещество |
ГОСТ 5903-77 |
- штучные торты: |
|||||||||||||
|
- жира в пересчете на сухое вещество |
до 200 г - 5 %, |
||||||||||||||
|
- сернистой кислоты |
200 - 250 г - 4 %, |
||||||||||||||
|
- сорбиновой кислоты |
ГОСТ 26181-84 и п. 3.7 ТУ |
250 - 500 г - 2,5 %, |
|||||||||||||
|
- сахарозы в водной фазе крема |
ГОСТ 5903-77 |
500 - 1000 г - 1,5 %, |
|||||||||||||
|
|
|
|
|
|
|
свыше 1000 г - 1,0 % |
|||||||||
|
|
|
|
|
|
|
- торты, изготавляемые на поточно-механизированных линиях, - 4 % |
|||||||||
|
|
|
|
|
|
|
- фасованные пирожные: |
|||||||||
|
|
|
|
|
|
|
до 500 г - 3 %, |
|||||||||
|
|
|
|
|
|
|
500 - 1000 - 1,5 % |
|||||||||
|
|
|
|
|
|
|
- штучные пирожные: |
|||||||||
|
|
|
|
|
|
|
до 45 г - 3 %, |
|||||||||
|
|
|
|
|
|
|
свыше 45 г - 5 %; |
|||||||||
|
|
|
|
|
|
|
- пирожные, изготовляемые на поточно-механизированных линиях, - 8 % |
|||||||||
|
печенье разное, реализуемое по массе, пряники |
Рецептуры сборников |
То же |
Не менее 400 г |
Не менее 100 г |
Размельчение в механическом размельчителе |
- |
- |
- |
- |
||||||
|
коврижки и бисквит, реализуемые по массе |
Рецептуры сборников |
От партии до 50 единиц транспортной тары - 3 единицы; от 51 до 150 единиц - 5 единиц, от 151 до 500 единиц - 8 единиц, от 501 до 1200 единиц - 13 единиц |
Не менее 1 шт. изделия, если масса изделия не превышает 400 г, если масса изделия более 400 г, то его разрезают на 4 равные части под прямым углом и отбирают одну четвертую часть |
Не менее 100 г |
- |
- |
- |
- |
- |
||||||
|
кексы, рулеты, ромовые бабы, реализуемые по массе |
То же |
То же |
То же |
Не менее половины изделия - при массе нетто свыше 1 кг; 1 шт. изделия при массе нетто 1 кг включительно |
Не менее 100 г |
- |
- |
- |
- |
- |
|||||
|
ромовые бабы, рулеты, кексы, сдобная пахлава и др. штучные изделия |
Рецептуры сборников |
От партии до 50 единиц транспортной тары - 3 единицы; от 51 до 150 единиц - 5 единиц, от 151 до 500 единиц - 8 единиц, от 501 до 1200 единиц - 13 единиц |
Не менее 1 шт. изделия, если масса изделия не превышает 150 г - отбирают не менее 400 г |
Не менее 100 г |
- |
- |
- |
- |
- |
||||||
|
22. |
Мороженое мягкое (сливочное и молочное 6 видов) |
ОСТ 282-77 |
5 % единиц транспортной тары при наличии в партии менее 20 единиц - отбирают одну |
Около 500 г |
Около 500 г |
Перемешивают путем перевертывания посуды с пробами не менее трех раз (при температуре (20 ± 2) °С |
Массовая доля: |
|
|
|
|||||
|
- жира |
|
|
|||||||||||||
|
- сахарозы |
|
|
|||||||||||||
|
- сухих веществ |
|
|
|||||||||||||
|
Кислотность |
ГОСТ 3624-67 |
|
|
||||||||||||
|
23. |
Полуфабрикаты и кулинарные изделия для диетического питания: |
|
|
|
|
|
|
|
|
|
|
||||
|
масло селедочное |
ТУ 28 РСФСР 03-78 |
От партии 2 - 25 единиц транспортной тары - 2 единицы; 26 - 90 - 3 единицы; 91 - 150 - 5 единиц; 151 - 280 - 8 единиц; 281 - 500 - 13 единиц и т.п. |
Не более 3 кг |
|
Тщательно перемешивают |
Массовая доля: |
|
|
|
||||||
|
- влаги |
|
|
|||||||||||||
|
- жира |
|
|
|||||||||||||
|
- поваренной соли |
|
|
|||||||||||||
|
паштет из печени |
ТУ 28 РСФСР 04-78 |
|
|
То же |
Массовая доля: |
|
|
|
|
||||||
|
- влаги |
|
|
|
||||||||||||
|
- жира |
|
|
|
||||||||||||
|
|
|
||||||||||||||
|
|
|
||||||||||||||
|
- поваренной соли |
|
|
|
||||||||||||
|
паштет особый |
ТУ 28 РСФСР 05-78 |
То же |
|
|
|
То же |
То же |
То же |
- |
|
|||||
|
сырные палочки |
ТУ 28 РСФСР 06-79 |
От партии до 50 мест - 3 единицы транспортной тары, от 51 до 150 мест - 5 шт., от 151 до 500 мест - 8 шт., от 501 до 1200 мест - 13 шт. |
Не менее 400 г |
Не менее 100 г |
Измельчают механическим измельчителем |
Массовая доля: |
|
|
|
||||||
|
- влаги |
- |
- |
|||||||||||||
|
- жира (в пересчете на сухое вещество) |
|
|
|||||||||||||
|
масло сырное |
ТУ 28 РСФСР 16-79 |
5 % единиц транспортной тары, при наличии в партии менее 20 единиц - отбирают одну |
По 50 г от каждой единицы транспортной тары |
Около 50 г |
Объединенную пробу помещают в водяную баню с температурой (30 ± 2) °С. При перемешивании нагревают до получения измельченной массы и выделяют пробу для анализа |
Массовая доля: |
|
- |
- |
||||||
|
- сухих веществ |
|
|
|||||||||||||
|
- жира |
|
|
|||||||||||||
|
фарш говяжий диетический |
ТУ 28 РСФСР 08-79 |
Не менее 10 % мест от партии, при наличии в партии менее 10 мест - 2 места |
|
|
Растирают в ступке до получения гомогенной массы |
Массовая доля: |
|
2 % мест от выборки, но не менее двух мест и выборочно проверяют по 10 порций из каждого места |
±2 % для одной порции, для 10 порций отклонения в меньшую сторону не допускаются |
||||||
|
- жира |
ОСТ 49 51-72 |
||||||||||||||
|
- влаги |
|||||||||||||||
|
полуфабрикаты рубленые повышенной пищевой ценности из говядины |
ТУ 28 РСФСР 01-78 |
ГОСТ 2488-76 |
10 % партии, если в партии меньше 10 мест - 2 места |
|
|
То же |
Массовая доля: |
|
То же |
±5 % для 1 шт. Масса 10 шт. изделий не должна иметь отклонений в меньшую сторону |
|||||
|
- сухих веществ |
|||||||||||||||
|
- поваренной соли |
ГОСТ 9957-79 |
||||||||||||||
|
- хлеба с учетом панировочных сухарей |
|||||||||||||||
|
- муки |
|||||||||||||||
|
полуфабрикат рубленый. Котлеты панированные в отрубях |
ТУ 28 РСФСР 29-90 |
То же |
10 шт. |
4 шт. массой 75 г и более или 6 шт. массой 50 г |
Вместе с панировкой растирают в ступке или дважды измельчают в мясорубке, перемешиваюи |
Массовая доля: |
|
|
2 % мест от выборки, но не меньше 2 мест. Проверяют по 10 порций из каждого места |
10 шт. ± 5 % |
|||||
|
- влаги |
|
10 шт. ± 2 % |
|||||||||||||
|
- хлеба с учетом панировки |
|||||||||||||||
|
- соли |
|
ГОСТ 9957-79 |
|||||||||||||
|
Масса полуфабриката |
|
Взвешиванием |
|||||||||||||
|
рулет мясной паровой, фаршированный омлетом |
ТУ 28 РСФСР 39-90 |
От партии до 10 единиц упаковок - 3 единицы, от 11 до 100 - 5 единиц, от 101 до 1000 - 10 единиц, от 1000 до 3000 - 15 единиц и т.д. |
|
|
|
Массовая доля: |
|
|
- |
- |
|||||
|
- начинки |
|
Взвешиванием |
|||||||||||||
|
- хлеба в основе |
|
||||||||||||||
|
- влаги в основе и начинке |
|
||||||||||||||
|
- поваренной соли |
|
||||||||||||||
|
полуфабрикаты рубленые из мяса кур (котлеты пожарские, кнели) |
ТУ 28 РСФСР 02-78 |
То же |
Не менее 10 % мест от партии, при наличии в партии менее 10 мест - 2 места |
10 шт. |
4 шт. массой 75 г и более или 6 шт. массой 50 г |
Растирают в ступке или дважды измельчают в мясорубке, перемешивают |
Массовая доля: |
|
|
2 % мест от выборки, но не меньше 2 мест. Выборочно проверяют по 10 порций из каждого места |
1 шт. ± 5 %, масса 10 шт. изделий не должна иметь отклонений в меньшую сторону |
||||
|
- влаги |
|
||||||||||||||
|
- хлеба с учетом панировки (для котлет) |
|||||||||||||||
|
- риса (для кнелей) |
|
ТУ 28 РСФСР 02-78 |
|||||||||||||
|
- поваренной соли |
|
||||||||||||||
|
Кислотность |
|
||||||||||||||
|
осетрина и севрюга отварные |
ТУ 28 РСФСР 09-79 |
Из партии от 2 до 25 единиц транспортной тары - 2 единцы, от 26 до 90 - 3 единицы, от 91 до 150 - 5 единиц, от 151 до 280 - 8 единиц, от 281 до 500 - 13 единиц и т.д. |
3 куска рыбы или 3 рыбы общей массой не более 0,6 кг. От фасованных кулинарных изделий отбирают не более трех единиц потребительской тары |
|
Пробу освобождают от несъедобных частей (хрящей). Плотную часть пропускают через мясорубку, смешивают с жидкой фракцией (при ее наличии) и растирают в ступке до однородной массы |
Массовая доля поваренной соли |
|
3 единицы неповрежденной потребительской тары от выборки. При разногласиях - 10 единиц неповрежденной потребительской тары от выборки и не более 0,03 % от общего количества в партии |
ТУ 28 РСФСР 09-79 |
||||||
|
рыба жареная «фри» |
ТУ 28 РСФСР 11-79 |
То же |
То же |
То же |
|
То же |
То же |
|
То же |
То же |
|
||||
|
Полуфабрикаты овощные кулинарные |
ТУ 28 РСФСР 07-79 |
ТУ 28 РСФСР 07-79 |
Не менее 10 % тарных единиц от партий, если в партии меньше 10 мест - 2 места |
300 г фарша или 3 шт. полуфабриката |
|
Дважды измельчают на мясорубке и тщательно перемешивают до получения однородной массы |
Массовая доля сухих веществ |
|
ГОСТ 4288-76 и п. 3.6 ТУ 28 РСФСР 07-79 |
2 % мест от выборки, но не менее 2 мест, выборочно проверяют по 10 порций из каждого места |
1 шт. ± 3 % |
||||
|
10 шт. ± 1 % |
|||||||||||||||
|
Изделия творожные кулинарные (пудинг творожный, зразы творожные с изюмом) |
ТУ 28 РСФСР 17-79 |
От партии до 50 единиц транспортной тары - 2 единицы, от 51 до 100 - 3 единицы, от 101 до 200 4 - единицы, от 201 до 300 - 5 единиц, от 301 и более - 6 единиц |
Около 500 г |
Около 100 г для пудинга, около 150 г для зраз |
Растирают в ступке, предварительно удалив наполнители (изюм) |
Массовая доля: |
|
|
10 шт. (зразы) |
± 3 % для 1 шт. изделия для 10 шт. - ± 1 % |
|||||
|
- сухих веществ |
|
ГОСТ 3628-73 |
|||||||||||||
|
- жира |
|
||||||||||||||
|
- сахара |
|
ГОСТ 3628-73 |
|||||||||||||
|
Общая кислотность |
|
ГОСТ 3624-67 |
|||||||||||||
|
Температура при выпуске с предприятия |
|
Измерением |
|||||||||||||
|
Полуфабрикаты крупяные (котлеты рисовые и манные) |
ТУ 28 РСФСР 13-79 |
ТУ 29 РСФСР 13-79 |
Не менее 10 % мест от партии, при наличии в партии менее 10 мест - 2 места |
2 порции полуфабрикатов |
|
Измельчают на мясорубке или тщательно перетирают в ступке, перемешивают |
Массовая доля: |
|
|
2 % мест от выборки, но не менее двух мест. Выборочно проверяют по 10 порций из каждого места |
То же |
||||
|
- сухих веществ |
|
||||||||||||||
|
- жира |
|
||||||||||||||
|
|
|
||||||||||||||
|
Изделия крупяные кулинарные (крупеник с творогом, пудинг рисовый) |
ТУ 28 РСФСР 19-79 |
ТУ 28 РСФСР 19-79 |
То же |
Не менее 300 |
|
То же |
Массовая доля: |
|
|
- |
|
||||
|
- сухих веществ |
|
||||||||||||||
|
- жира |
|
||||||||||||||
|
- сахарозы |
|
||||||||||||||
|
Плов фруктовый |
ТУ 28 РСФСР 14-79 |
ТУ 28 РСФСР 14-79 |
Не менее 10 % мест от партии, при наличии в партии менее 10 мест - 2 места |
Не менее 500 г |
|
То же |
Массовая доля: |
|
|
- |
|
||||
|
- сухих веществ в рисовой фазе |
|
||||||||||||||
|
- жира в рисовой фазе |
|
||||||||||||||
|
- фруктов |
|
Взвешиванием |
|||||||||||||
|
Тесто для вареников ленивых (жирное и полужирное) |
ТУ 28 РСФСР 18-79 |
|
|
|
|
Массовая доля: |
|
|
|
|
|||||
|
- жира |
|
||||||||||||||
|
- сухих веществ |
|
||||||||||||||
|
- сахарозы |
|
||||||||||||||
|
Кислотность |
|
ГОСТ 3624-67 |
|||||||||||||
|
Температура при выпуске с предприятия |
|
Измерением |
|||||||||||||
|
Кремы творожно-фруктовые |
ТУ 28 РСФСР 15-79 |
То же |
|
|
|
|
Массовая доля: |
|
|
10 порций |
±3 % для одной порции, ±1 % для 10 порций |
||||
|
- сухих веществ |
|
||||||||||||||
|
- сахарозы |
|
||||||||||||||
|
Кислотность |
|
ГОСТ 3624-67 |
|||||||||||||
|
Температура при выпуске с предприятия |
|
Измерением |
|||||||||||||
|
Изделия сладкие кулинарные (желе, самбуки, муссы, кремы сливочные) |
ТУ 28 РСФСР 20 -79 |
То же |
|
|
|
|
Массовая доля: |
|
|
10 порций |
1 порция - ±3 % |
||||
|
- сухих веществ |
|
10 порций - ±1 % |
|||||||||||||
|
|
|
ГОСТ 8756.2-82 |
|||||||||||||
|
- сахарозы в желе, кремах |
|
||||||||||||||
|
Кислотность в желе, кремах |
|
ГОСТ 3624-67 |
|||||||||||||
|
Температура при выпуске с предприятия |
|
Измерением |
|||||||||||||
|
Яблоко в тесте |
ТУ 28 РСФСР 21-79 |
|
|
6 шт. |
|
Массовая доля: |
|
|
10 штук |
Не более (-5 %) для 1 шт., ±4 % для 10 шт. |
|||||
|
- начинки |
|
Взвешиванием |
|||||||||||||
|
- сахара в основе |
|
||||||||||||||
|
- жира в основе |
|
|
|||||||||||||
|
- влаги в основе |
|
||||||||||||||
|
Пирожки из дрожжевого теста |
ТУ 28 РСФСР 22-79 |
То же |
|
|
|
|
Массовая доля: |
|
То же |
То же |
То же |
||||
|
- начинки |
|
||||||||||||||
|
- сахара |
|
||||||||||||||
|
- жира |
|
||||||||||||||
|
- влаги (основы и начинки) |
|
||||||||||||||
|
Пирожки слоеные |
ТУ 28 РСФСР 23-79 |
|
|
|
|
Массовая доля: |
|
|
10 шт. |
Не более (-5 %) для 1 шт., ±4 % для 10 шт. |
|||||
|
- начинки |
|
|
|||||||||||||
|
- влаги (основы и начинки) |
|
||||||||||||||
|
Ватрушки с повидлом |
ТУ 28 РСФСР 24-79 |
То же |
|
|
6 шт. |
|
Массовая доля: |
|
|
То же |
То же |
||||
|
- влаги (основы) |
|
||||||||||||||
|
- начинки |
|
||||||||||||||
|
- сахара в основе |
|
||||||||||||||
|
- жира |
|
||||||||||||||
Приложение 2
Отбор проб блюд и кулинарных изделий и исследуемые физико-химические показатели качества
|
№ п/п |
Группа блюд (изделий) |
Кол-во блюд (порций изделий) |
Показатели |
Методы анализа и источники |
|||||
|
для определения средней массы порций или изделия |
для физико-химического анализа |
||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
||||
|
1. |
Холодные блюда: |
|
|
Определение содержания: |
|
||||
|
салаты из свежих овощей |
3 порции |
2 порции (не менее 200 г) |
жира |
Экстракционный по обезжиренному остатку, метод Гербера, экстракционно-весовой (ускоренный) |
|||||
|
винегрет овощной |
3 |
1 - 2 (не менее 200 г) |
сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой |
|||||
|
Определение содержания: |
|
||||||||
|
жира |
Метод Гербера, экстракционно-весовой |
||||||||
|
винегрет с добавкам |
3 |
1 - 2 (не менее 200 г) |
Масса добавки (сельди, мяса и т.д.) |
Взвешивание |
|||||
|
Определение содержания: |
|
||||||||
|
жира |
Метод Гербера, экстракционно-весовой |
||||||||
|
сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой |
||||||||
|
салаты мясные и рыбные |
3 |
1 - 2 (не менее 200 г) |
Масса мяса |
Взвешивание |
|||||
|
Определение содержания: |
|
||||||||
|
сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой |
||||||||
|
жира |
Метод Гербера, экстракционно-весовой |
||||||||
|
студни |
3 |
2 |
Масса плотной части |
Взвешивание |
|||||
|
блюда заливные |
3 |
2 |
Масса основного изделия (мяса, рыбы) |
Взвешивание |
|||||
|
паштеты, масло селедочное |
3 |
1 - 2 (не менее 100 г) |
Определение содержания: |
|
|||||
|
сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой |
||||||||
|
жира |
Метод Гербера, экстракционно-весовой |
||||||||
|
сельдь рубленая |
|
|
Определение содержания: |
|
|||||
|
сухих веществ |
Высушивание в сушильном шкафу при 130 °С |
||||||||
|
бутерброды |
10 |
- |
хлеба |
Любой химический метод по МУ |
|||||
|
2. |
Супы: |
|
|
|
|
||||
|
заправочные без мяса, птицы, рыбы |
3 |
2 - 3 (не менее 300 г) |
Определение содержания: |
|
|||||
|
сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой после выпаривания жидкости |
||||||||
|
жира |
Метод Гербера, экстракционно-весовой |
||||||||
|
супы с мясом, птицей, рыбой, фрикадельками, клецками, гренками |
10 |
- |
Масса мяса, птицы, рыбы, фрикаделек, клецек, гренок |
|
|||||
|
Определение содержания: |
|
||||||||
|
сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой |
||||||||
|
жира |
Метод Гербера, экстракционно-весовой |
||||||||
|
Солянки (мясная сборная, рыбная) и холодные супы |
3 |
2 - 3 (не менее 300 г) |
Масса мясных продуктов*(1) Определение содержания жира (в жидкой части) |
Взвешивание. Метод Гербера, экстракционно-весовой |
|||||
|
супы-пюре из разных овощей |
3 |
2 - 3 (не менее 300 г) |
Определение содержания: |
|
|||||
|
сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой |
||||||||
|
жира |
Метод Гербера или экстракционно-весовой |
||||||||
|
бульон с гарниром |
3 |
2 - 3 (не менее 300 г) |
Масса гарнира |
Взвешивание. Высушивание в сушильном шкафу |
|||||
|
Определение содержания сухих веществ (в жидкой части) |
|
||||||||
|
молочные с макаронными изделиями или крупой |
3 |
2 - 3 (не менее 300 г) |
Определение содержания: |
|
|||||
|
сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой |
||||||||
|
жира |
Метод Гербера, экстракционно-весовой |
||||||||
|
Определение содержания молока (по лактозе) |
Перманганатный, ускоренный цианидный |
||||||||
|
сладкие супы с фруктами, гарниром и сметаной*(1а) |
3 для определения плотной части необходимо 5 порций |
2 - 3 (не менее 300 г) |
Масса плотной части. Определение содержания общего сахара |
Взвешивание. Любой химический метод по МУ |
|||||
|
3. |
Блюда из рыбы: |
|
|
|
|
||||
|
отварной, припущенной, тушеной, жареной рыбы; жареной, панированной в муке и сухарях, с гарниром и жиром или соусом |
3 |
2 (не менее 100 г) |
Масса основного изделия |
Взвешивание |
|||||
|
Определение массы панировки и выхода рыбы*(2) |
Взвешивание |
||||||||
|
Определение достаточности термической обработки |
Проба на пероксидазу, фосфатазу |
||||||||
|
основное изделие |
10 |
- |
|
|
|||||
|
гарнир с жиром*(3) или соусом*(4) |
|
200 г |
Определение содержания: |
|
|||||
|
сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой |
||||||||
|
жира |
Метод Гербера, экстракционно-весовой |
||||||||
|
из котлетной массы (биточки, котлеты, рулет, тефтели с гарниром и жиром или соусом) |
3 |
2 |
Масса основного изделия |
Взвешивание |
|||||
|
Основное изделие |
10 |
4 - при массе 75 г и более; 6 - при массе 50 г |
Определение содержания: |
|
|||||
|
сухих веществ*(5) |
Высушивание в сушильном шкафу |
||||||||
|
хлеба |
Йодометрический, цианидный |
||||||||
|
Определение достаточности термической обработки |
Проба на пероксидазу, фосфатазу |
||||||||
|
гарнир с жиром или соусом |
См. выше |
|
|
|
|||||
|
биточки, котлеты рыбные (без хлеба) с гарниром и жиром |
3 |
2 |
Масса основного изделия |
Взвешивание |
|||||
|
основное изделие |
10 |
4 - при массе 75 г и более; 6 - при массе 50 г |
Определение содержания сухих веществ |
Высушивание в сушильном шкафу |
|||||
|
Качественное определение наполнителя |
Реакция на присутствие хлеба с помощью реактива Люголя |
||||||||
|
Определение достаточности термической обработки |
Проба на пероксидазу, фосфатазу |
||||||||
|
гарнир с жиром |
|
См. выше |
|
|
|||||
|
4. |
Блюда из мяса и мясных продуктов, птицы и кролика: |
|
|
|
|
||||
|
из отварного, тушеного мяса и мясных продуктов с гарниром и соусом |
3 |
2 |
Масса основного изделия |
Взвешивание |
|||||
|
гарнир с соусом |
|
См. выше |
|
|
|||||
|
из жареного мяса (натуральные порционные, жареные фри) с жиром и гарниром |
3 |
2 |
|
|
|||||
|
основное изделие |
Взвешивают не менее 10 порций |
- |
Определение массы панировки и выхода мяса |
Взвешивание |
|||||
|
Определение достаточности термической обработки |
Проба на пероксидазу, фосфатазу |
||||||||
|
Качество фритюрного жира |
Методы определения качества фритюра |
||||||||
|
гарнир с жиром |
|
См. выше |
|
|
|||||
|
из котлетной массы (котлеты, биточки, шницели, тефтели) с гарниром и жиром или соусом |
3 |
2 |
Масса основного изделия |
Взвешивание |
|||||
|
основное изделие |
10 |
массой 75 г и более - 4 изд., массой 50 г - 6 изд. |
Определение содержания: |
Высушивание в сушильном шкафу |
|
ГОСТ 4298-76 |
|||
|
сухих веществ*(7) хлеба |
Йодометрический, цианидный |
||||||||
|
соли |
|||||||||
|
Определение: |
|
||||||||
|
достаточности термической обработки |
Проба на пероксидазу, фосфатазу |
||||||||
|
наличия добавок субпродуктов |
Люминесцентный, гистологический |
||||||||
|
содержание сухожилий |
Люминесцентный |
||||||||
|
гарнир с жиром или соусом |
См. выше |
|
|
|
|||||
|
зразы, рулеты с гарниром и соусом |
3 |
2 |
Масса основного изделия |
Взвешивание |
|||||
|
основное изделие |
10 (4 изделия) |
|
Определение достаточности термической обработки |
Проба на пероксидазу, фосфатазу |
|||||
|
из тушеного жареного мяса, приготовленного с соусом/азу, поджарка, бефстроганов, гуляш, рагу/, с гарниром |
2 |
2 (не менее 200 г) |
Масса основного изделия |
Взвешивание |
|||||
|
Определение содержания: |
|
||||||||
|
сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой |
||||||||
|
жира |
Метод Гербера, экстракционно-весовой |
||||||||
|
5. |
Блюда из рубленого мяса: |
|
|
|
|
||||
|
натуральные /бифштекс, котлеты, шницели/ с гарниром и жиром |
3 |
2 |
Масса основного изделия |
Взвешивание |
|||||
|
основное изделие |
|
4 шт. |
Качественное определение наполнителя |
Реакция на присутствие хлеба с помощью раствора Люголя |
|
||||
|
Определение содержания сухих веществ |
Высушивание в сушильном шкафу |
||||||||
|
Определение содержания соли |
|||||||||
|
Определение достаточности термической обработки |
Проба на пероксидазу, фосфатазу |
||||||||
|
Определение наличия добавок субпродуктов |
Люминесцентный, гистологический |
||||||||
|
Определение содержания сухожилий |
Люминесцентный |
||||||||
|
гарнир с жиром |
|
См. выше |
|
См. выше |
|||||
|
Определение содержания: |
|
||||||||
|
фарша |
Взвешивание |
||||||||
|
хлеба в мясной оболочке |
Йодометрический, цианидный - ГОСТ 4288-76 |
||||||||
|
гарнир с соусом |
1 |
См. выше |
|
|
|||||
|
голубцы, кабачки, помидоры, баклажаны, фаршированные мясом, с соусом |
2 |
2 |
Масса основного изделия |
Взвешивание |
|||||
|
Определение содержания: |
|
||||||||
|
сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой |
||||||||
|
жира |
Метод Гербера, экстракционно-весовой |
||||||||
|
6. |
Блюда из картофеля, овощей, грибов и бобовых: |
|
|
|
|
||||
|
отварные, припущенные, тушеные, жареные, запеченные, заправленные жиром, сметаной или соусом |
3 |
2 (не менее 200 г) |
Определение содержания: |
|
|||||
|
сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой |
||||||||
|
жира |
Метод Гербера, экстракционно-весовой |
||||||||
|
запеканки, пудинги, фаршированные овощи, овощные котлеты, зразы, рулет с жиром, сметаной или соусом |
То же |
2 (не менее 200 г) |
Определение содержания: |
|
|||||
|
сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой |
||||||||
|
жира*(8) |
Метод Гербера, экстракционно-весовой |
||||||||
|
основное изделие |
10 (порционируемых изделий) |
1 (порционируемое изделие |
Определение содержания: |
|
|||||
|
сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой |
||||||||
|
жира*(9) |
Метод Гербера, экстракционно-весовой |
||||||||
|
Наличие яиц |
Качественная реакция |
||||||||
|
7. |
Блюда из круп и макаронных изделий: |
|
|
|
|
||||
|
каши рассыпчатые, макароны или лапша отварные |
|
200 г |
Определение содержания: |
|
|||||
|
сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой |
||||||||
|
жира |
Метод Гербера, экстракционно-весовой |
||||||||
|
каши молочные жидкие и вязкие |
|
|
Свежесть*(10) |
Метод определения кислотности |
|||||
|
Определение содержания: |
|
||||||||
|
сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой |
||||||||
|
жира |
Метод Гербера, экстракционно-весовой |
||||||||
|
молока (по лактозе) |
Перманганатный, ускоренный, цианидный |
||||||||
|
котлеты, биточки, запеканки, крупеник, макаронник, лапшевник с жиром, сметаной или соусом |
3 |
2 (не менее 200 г) |
Определение содержания: |
|
|||||
|
сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой |
||||||||
|
жира |
Метод Гербера, экстракционно-весовой |
||||||||
|
основное изделие: |
|
|
|
|
|||||
|
котлеты, биточки |
|
|
|
|
|||||
|
запеканки, крупеник, макаронник, лапшевник |
10 |
3 |
Определение содержания: |
|
|||||
|
сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой |
||||||||
|
10 |
2 (не менее 200 г) |
жира |
Метод Гербера, экстракционно-весовой |
||||||
|
молока (по лактозе)*(11) |
Перманганатный, ускоренный, цианидный |
||||||||
|
сахара*(12) |
Любой химический метод по МУ |
||||||||
|
Наличие яиц |
Качественная реакция |
||||||||
|
8. |
Блюда из яиц: |
|
|
|
|
||||
|
омлеты с жареным картофелем, луком, морковью и другими продуктами |
|
|
Определение содержания: |
|
|||||
|
сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой |
||||||||
|
жира |
Метод Гербера, экстракционно-весовой |
||||||||
|
9. |
Блюда из творога: |
|
|
|
|
||||
|
сырники, пудинги, запеканки со сметаной или соусом |
3 |
2 (не менее 200 г) |
Определение содержания: |
|
|||||
|
сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой |
||||||||
|
жира |
Метод Гербера, экстракционно-весовой |
||||||||
|
основное изделие |
10 (порционируемых изделий) |
2 (порционируемых изделий |
Кислотность |
Метод определения кислотности |
|||||
|
Определение содержания: |
|
||||||||
|
сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой |
||||||||
|
жира |
Метод Гербера, экстракционно-весовой |
||||||||
|
сахара*(14) |
Любой химический метод по МУ |
||||||||
|
муки*(15) |
Определение содержания крахмала муки |
||||||||
|
Наличие яиц |
Качественная реакция |
||||||||
|
10. |
Мучные блюда: |
|
|
|
|
||||
|
пельмени, вареники с жиром или сметаной |
3 |
2 |
Определение содержания: |
|
|||||
|
сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой |
||||||||
|
жира |
Метод Гербера, экстракционно-весовой |
||||||||
|
основное изделие |
|
|
Исследуют полуфабрикат, как указано в табл. |
|
|||||
|
блинчики с разными фаршами, с жиром, сметаной |
3 |
1 - 2 |
Определение содержания: |
|
|||||
|
сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой |
||||||||
|
жира |
Метод Гербера, экстракционно-весовой |
||||||||
|
основное изделие |
10 изделий |
Блинчики 3 изделия |
Определение содержания: |
|
|||||
|
фарша*(16) |
Взвешивание |
||||||||
|
сухих веществ в фарше |
Высушивание в сушильном шкафу или в приборе Чижовой |
||||||||
|
сухих веществ (в оболочке) |
Высушивание в сушильном шкафу |
||||||||
|
Наличие яиц (в оболочке) |
Качественная реакция |
||||||||
|
блины с маслом, сметаной и другими продуктами |
3 |
2 |
Определение содержания: |
|
|||||
|
сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой |
||||||||
|
жира |
Метод Гербера, экстракционно-весовой |
||||||||
|
основное изделие |
10 изделий |
Блины массой 50 г - 4 изделия |
Определение содержания сухих веществ |
Высушивание в сушильном шкафу |
|||||
|
Кислотность |
Метод определения кислотности |
||||||||
|
Влажность |
Высушивание в сушильном шкафу - ТУ 28.2-79 |
||||||||
|
оладьи, вырабатываемые на машине типа МПО.400 |
3 |
2 |
Наличие яиц |
Качественная реакция - ТУ 28.2-79 |
|||||
|
Кислотность |
Метод определения кислотности - ГОСТ 5670-51 |
||||||||
|
Определение содержания: |
|
||||||||
|
сахара*(17) |
Перманганатный, ускоренный |
||||||||
|
жира в мякише*(18) |
Йодометрический - ТУ 28.2-79 |
||||||||
|
|
Рефрактометрический - ТУ 28.2-79 |
||||||||
|
оладьи, вырабатываемые вручную, с маслом, сметаной и другими продуктами |
3 |
2 |
Определение содержания: |
|
|||||
|
сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой |
||||||||
|
жира |
Метод Гербера, экстракционно-весовой |
||||||||
|
основное изделие |
10 изделий |
Оладьи массой 75 г - 4 изделия |
Определение содержания сухих веществ |
Высушивание в сушильном шкафу |
|||||
|
Наличие яиц |
Качественная реакция |
||||||||
|
11. |
Гарниры: |
|
|
|
|
||||
|
из картофеля, овощей, бобовых, круп и макаронных изделий |
|
200 г |
Определение содержания: |
|
|||||
|
сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой |
||||||||
|
жира |
Метод Гербера, экстракционно-весовой |
||||||||
|
12. |
Соусы (кроме молочных): |
|
|
Определение содержания: |
|
||||
|
сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой |
||||||||
|
жира |
Метод Гербера, экстракционно-весовой |
||||||||
|
молочный |
|
|
Определение содержания: |
|
|||||
|
сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой |
||||||||
|
молока (по лактозе) |
Перманганатный, ускоренный, цианидный |
||||||||
|
жира |
Метод Гербера, экстракционно-весовой |
||||||||
|
молочный сладкий |
|
|
Определение содержания: |
|
|||||
|
сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой |
||||||||
|
сахара*(18а) |
Любой химический метод по МУ |
||||||||
|
молока (по лактозе) |
Перманганатный, ускоренный, цианидный |
||||||||
|
13. |
Сладкие блюда: |
|
|
|
|
||||
|
компоты, фруктовоягодные кисели |
3 |
2 (не менее 300 г) |
Масса плотной части (компоты) |
Взвешивание |
|||||
|
Определение содержания |
|
||||||||
|
сахара (в киселях) |
Рефрактометрический |
||||||||
|
сухих веществ |
Рефрактометрический |
||||||||
|
желе, кремы, муссы (без крупы), самбуки с сахаром или соусом |
То же |
2 (не менее 200 г) |
Определение содержания: |
|
|||||
|
сухих веществ |
Высушивание в сушильном шкафу или рефрактометрический (кроме кремов) |
||||||||
|
сахара жира (в кремах) |
Рефрактометрический, Метод Гербера, экстракционно-весовой |
||||||||
|
муссы с манной крупой с сиропом |
То же |
|
Определение содержания: |
|
|||||
|
сухих веществ |
Высушивание в сушильном шкафу |
||||||||
|
сахара |
Рефрактометрический |
||||||||
|
основное изделие |
10 порционируемых изделий |
2 порционируемых изделия |
Определение содержания: |
|
|||||
|
сухих веществ |
Высушивание в сушильном шкафу |
||||||||
|
сахара |
Рефрактометрический |
||||||||
|
манной крупы*(19) |
Любой химический метод по МУ |
||||||||
|
сироп |
- |
100 г |
Определение содержания сухих веществ |
Рефрактометрический |
|||||
|
Кисель и желе молочные |
3 |
2 (не менее 300 г) |
Определение содержания: |
|
|||||
|
сухих веществ |
Высушивание в сушильном шкафу или рефрактометрический |
||||||||
|
сахара |
Рефрактометрический |
||||||||
|
молока (по лактозе) |
Перманганатный, ускоренный, цианидный |
||||||||
|
выпеченные сладкие блюда (пудинг, шарлотка и т.д.) |
3 |
1 (не менее 200 г) |
Масса основного изделия |
Взвешивание |
|||||
|
с сиропом или соусом |
|
|
Определение содержания: |
|
|||||
|
сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой |
||||||||
|
жира |
Метод Гербера, экстракционно-весовой |
||||||||
|
общего сахара |
Любой химический метод по МУ |
||||||||
|
Основное изделие |
10 |
1 |
Содержание сухих веществ |
Высушивание в сушильном шкафу или в приборе Чижовой |
|||||
|
Содержание жира |
Метод Гербера, экстракционно-весовой |
||||||||
|
Содержание сахара |
Любой химический метод по МУ |
||||||||
|
14. |
Горячие напитки: |
|
|
|
|
||||
|
чай, кофе черный с сахаром |
3 порции |
2 (не менее 300 г) |
Содержание сухих веществ |
Рефрактометрический |
|||||
|
Содержание сахара |
Рефрактометрический |
||||||||
|
Определение полноты вложения сырья (кофе) в кофе черный |
Колориметрический или фотометрический |
||||||||
|
кофе с молоком, какао с молоком |
|
2 (не менее 300 г) |
Содержание сухих веществ |
Рефрактометрический*(20) |
|||||
|
Содержание сахара |
Рефрактометрический |
||||||||
|
Содержание молока (по лактозе) |
Перманганатный, ускоренный, цианидный |
||||||||
|
Определение полноты вложения сырья (кофе) в кофе с молоком |
Фотометрический |
||||||||
|
молоко кипяченое |
|
2 порции |
Определение плотности |
ГОСТ 3625-71 |
|||||
|
Содержание лактозы |
Перманганатный, ускоренный, цианидный |
||||||||
|
Содержание сухих веществ (экстракта) |
Рефрактометрический |
||||||||
|
Определение полноты вложения сырья (кофе) |
Колориметрический или фотометрический |
||||||||
|
чай-настой и напиток без сахара |
|
1 порция 55 мл*(22) |
Определение: |
|
|||||
|
содержания экстрактивных веществ (в настое и напитке) |
Высушивание в сушильном шкафу |
||||||||
|
полноты вложения сырья, содержания танина |
Колориметрический ГОСТ 19885-74. Качественная реакция |
||||||||
|
Обнаружение жженого сахара |
|
||||||||
|
15. |
Холодные напитки: из плодов, ягод, фирменные |
3 |
2 |
Свежесть настоя чая |
То же |
||||
|
Определение содержания: |
|
||||||||
|
коктейли молочные |
2*(23) |
2*(24) |
сухих веществ жира |
Рефрактометрический, Метод Гербера - ГОСТ 5867-69 |
|||||
|
сухих веществ |
Высушивание в сушильном шкафу - ГОСТ 3626-73 |
||||||||
|
Коктейли алкогольные |
2*(25) |
2*(24) |
Масса наполнителя |
Взвешивание |
|||||
|
Определение содержания спирта |
Метод определения содержания спирта - ГОСТ 13191-73, с указаниями в части подготовки фильтрата |
||||||||
|
Определение содержания общего экстракта |
Метод определения содержания общего экстракта рефрактометрический с предварительной отгонкой спирта |
||||||||
_________________
*(1) Потери массы при тепловой обработке мясных продуктов, внесенных в готовое блюдо, см. раздел "Отбор и подготовка блюд".
*(1а) В сладких супах с гарниром, свежими фруктами и сметаной определяют содержание жира.
*(2) Панированных в муке и сухарях.
*(3) Здесь и далее подразумевать жир, используемый для заправки гарнира.
*(4) Здесь и далее дополнительно последуют контрольные гарнир и соус, см. раздел "Отбор проб".
*(5) Определяют в полуфабрикате тефтелей.
*(6) В панированных изделиях.
*(7) Определяют в полуфабрикате тефтелей (в связи с тушением изделия).
*(8), *(9) В изделиях, в рецептуру которых входят жиросодержащие продукты.
*(10), *(12) В случае сомнения при органолептческой оценке.
*(11) В изделиях, в рецептуру которых входит молоко.
*(13) Здесь и далее основное изделие отбирают только в случае сомнения при органолептической оценке.
*(14) В случае сомнения при органолептической оценке.
*(15) Количество редуцирующих сахаров до инверсии, после инверсии (общего сахара) и после полного гидролиза углеводов определяют обязательно одним методом.
*(16) Кроме блинчиков с творогом (определяют в полуфабрикате).
*(17) Определяют в спорных (арбитражных) случаях.
*(18) Содержание жира определяют в мякише сдобных оладий с молоком.
*(18а) Кол-во редуцирующих сахаров до инверсии, после инверсии (общего сахара) определяют обязательно одним методом.
*(19) Кол-во редуцирующих сахаров после инверсии (общего сахара) и после полного гидролиза углеводов определяют обязательно одним методом.
*(20) При расчете содержания сухих в-в в кофе и какао с молоком по формуле определяют также содержание сухих в-в в пастеризованном молоке.
*(21) Порции, отбираемые методом контрольной закупки или у потребителя.
*(22) Заварку (настой) из чайника.
*(23) Порции, взятые для взвешивания методом контрольной закупки или у потребителя.
*(24) Параллельно готовят 2 порции (эталон).
Приложение 3
Основные физико-химические показатели качества пищевых продуктов, предусмотренные ГОСТами
|
№ п/п |
Наименование продуктов |
Стандарты на продукты* |
Физико-химические показатели по ГОСТам |
ГОСТы на методы испытаний* |
||||
|
1 |
2 |
3 |
4 |
5 |
||||
|
1. |
МЯСО И МЯСОПРОДУКТЫ |
|
||||||
|
Мясо: |
|
|
|
|||||
|
говядина и телятина |
ГОСТ 779-87 |
Свежесть |
|
|||||
|
|
|
|
|
ГОСТ 19496-74 |
||||
|
|
|
Пестициды |
|
По методам, утвержденным Минздравом СССР |
||||
|
баранина и козлятина в тушах |
Свежесть |
|
||||||
|
свинина в тушах и полутушах |
Масса, температура |
|
||||||
|
|
|
Свежесть |
|
ГОСТ 19496-74, |
||||
|
|
|
|
|
|||||
|
блоки мясные |
Доброкачественность |
|
||||||
|
замороженные блоки из жилованного мяса и субпродуктов |
ОСТ 10-02-0104-86 |
Масса блоков |
|
|
||||
|
|
|
Температура в центре блока |
|
|
||||
|
замороженные |
|
Свежесть |
|
|||||
|
|
|
|
|
ГОСТ 19496-74 |
||||
|
Мясные продукты: |
|
|
|
|
||||
|
продукты из свинины запеченные и жареные (буженина, карбонат, шейка московская) |
Массовая доля поваренной соли, % |
|
||||||
|
продукты из свинины сырокопченые |
Массовая доля поваренной соли, % |
|
||||||
|
|
|
Массовая доля нитрата |
|
|||||
|
продукты из свинины вареные |
Массовая доля: |
|
|
|||||
|
|
|
- поваренной соли |
|
|||||
|
|
|
- нитрата |
|
|||||
|
|
|
Остаточная активность кислой фосфатазы |
|
|||||
|
продукты из свинины копчено-вареные |
Массовая доля: |
|
|
|||||
|
- поваренной соли |
|
|||||||
|
- нитрита |
|
|||||||
|
продукты из свинины копчено-запеченные |
То же |
|
То же |
|||||
|
Колбасные изделия: |
|
|
|
|
||||
|
колбасы сырокопченые |
Массовая доля: |
|
|
|||||
|
- влаги |
|
|||||||
|
- поваренной соли |
|
|||||||
|
- нитрита |
|
|||||||
|
колбасы варено-копченые |
То же |
|
То же |
|||||
|
колбасы полукопченые |
Массовая доля: |
|
|
|||||
|
- влаги |
|
|||||||
|
- поваренной соли |
|
|||||||
|
- нитрита |
|
|||||||
|
паштет печеночный |
Массовая доля: |
|
|
|||||
|
- поваренной соли |
|
ГОСТ 8756.20-70 |
||||||
|
- жира |
|
|||||||
|
- солей олова, свинца |
|
|||||||
|
|
|
|||||||
|
Посторонние примеси |
|
|||||||
|
консервы мясорастительные "Каша с мясом" |
Массовая доля: |
|
|
|||||
|
- жира |
|
|||||||
|
- белка |
|
|||||||
|
- поваренной соли |
|
|||||||
|
- тяжелых металлов |
|
|||||||
|
|
|
|||||||
|
|
|
|||||||
|
Посторонние примеси |
|
Визуально |
||||||
|
изделия макаронные с мясом |
Массовая доля: |
|
|
|||||
|
- мяса |
|
|||||||
|
- жира |
|
|||||||
|
- хлоридов |
|
|||||||
|
Минеральные примеси |
|
|||||||
|
Примеси растительного происхождения |
|
|||||||
|
Посторонние примеси |
|
|||||||
|
Токсичные элементы |
|
|||||||
|
|
|
|||||||
|
|
|
ГОСТ 27935-86 |
||||||
|
колбасы вареные, сосиски и сардельки, хлебы мясные |
То же и массовая доля крахмала (для колбас, мясных хлебов) |
|
То же и ГОСТ 10574-73 |
|||||
|
Консервы мясные, мясорастительные: |
|
|
|
|
||||
|
говядина тушеная |
Массовая доля: |
|
|
|||||
|
- мяса и жира в % к массе нетто |
|
|||||||
|
- жира |
|
|||||||
|
- поваренной соли |
|
ГОСТ 8756.20-70 |
||||||
|
- солей олова, свинца |
|
|||||||
|
|
|
|||||||
|
Посторонние примеси |
|
|||||||
|
гуляш |
Массовая доля: |
|
|
|||||
|
- соуса к массе нетто |
|
|||||||
|
- мяса к массе нетто |
||||||||
|
- поваренной соли |
|
ГОСТ 8756.20-70 |
||||||
|
- солей олова, меди, свинца |
|
|||||||
|
|
|
|||||||
|
|
|
|||||||
|
Мясо птицы: тушки кур, уток, гусей, индеек, цесарок мясо цыплятбройлеров |
ГОСТ 21784-78 |
Свежесть |
|
|||||
|
То же |
|
То же |
||||||
|
2. |
ЖИРЫ: |
|
|
|
|
|||
|
масло коровье |
ГОСТ 37-87 |
|
|
|
||||
|
жиры животные топленые пищевые |
Массовая доля: |
|
|
|||||
|
- влаги |
|
ГОСТ 8285-74 |
||||||
|
- антиокислителей |
|
|||||||
|
Кислотное число |
|
|
||||||
|
Прозрачность (в растопленном состоянии, в единицах шкалы фотоэлектроколориметра) |
|
|
||||||
|
жиры кондитерские и кулинарные |
ОСТ 18-197-84 |
Массовая доля: |
|
|
||||
|
- жира |
|
|
||||||
|
- влаги |
|
|
||||||
|
Кислотное число |
|
|||||||
|
Температуры плавления, застывания |
||||||||
|
Твердость по Каминскому |
||||||||
|
жиры кондитерские и кулинарные |
|
Температура полного расплавления твердого жира для кондитерских изделий на основе какао-порошка, для жировой глазури, пралиновых масс и начинок для них |
ОСТ 18-197-84 |
|||||
|
Твердость по Воларовичу |
|
|
||||||
|
маргарин |
Массовая доля: |
|
|
|||||
|
- жира |
|
|
||||||
|
- влаги и летучих веществ |
|
|
||||||
|
- поваренной соли |
|
|
||||||
|
- твердых триглицеридов |
|
|||||||
|
Температура плавления жира, выделенного из маргарина |
||||||||
|
Кислотность |
|
|
||||||
|
Стойкость жидкого маргарина |
|
|
||||||
|
Твердость |
|
|
||||||
|
ЯЙЦА И ПРОДУКТЫ ИХ ПЕРЕРАБОТКИ: |
|
|
|
|
||||
|
яйца куриные пищевые |
Чистота скорлупы |
|
Визуально |
|||||
|
Масса яиц |
|
|||||||
|
Величина воздушной камеры, состояние белка, желтка и целостность скорлупы |
Просвечивание на овоскопе |
|||||||
|
порошок яичный |
Массовая доля: |
|
|
|||||
|
- влаги |
|
|||||||
|
- белковых веществ |
||||||||
|
- жира |
||||||||
|
- золы |
|
|
||||||
|
Растворимость |
|
|
||||||
|
Кислотность |
|
|
||||||
|
4. |
МОЛОКО И МОЛОЧНЫЕ ПРОДУКТЫ: |
|
|
|
|
|||
|
молоко коровье пастеризованное |
Плотность |
|
||||||
|
Кислотность |
|
ГОСТ 3624-67 |
||||||
|
Степень чистоты |
|
|||||||
|
Температура |
|
|||||||
|
Наличие фосфатазы |
|
|||||||
|
Массовая доля жира |
|
|||||||
|
молоко стерилизованное |
ОСТ 49 140-85 |
То же, кроме наличия фосфатазы |
|
То же, кроме ГОСТ 3623-73 |
||||
|
молоко витаминизированное для детей |
ОСТ 49 22-71 |
Массовая доля: |
|
|
||||
|
- жира |
|
|||||||
|
- сухого обезжиренного остатка |
|
|||||||
|
- витаминов (A, Д2, C) |
|
|||||||
|
Кислотность |
|
ГОСТ 3624-67 |
||||||
|
Степень чистоты по эталону |
|
|||||||
|
Температура |
|
|||||||
|
сливки из коровьего молока |
ТУ 10-02.02.789-08-89 |
|
|
|
||||
|
сметана |
ТУ 10-02.02.789-09-89 |
|
|
|
||||
|
кефир |
ОСТ 49 29-84 |
|
|
|
||||
|
творог мягкий диетический |
ОСТ 49 25-85 |
Массовая доля: |
|
|
||||
|
- жира |
|
|||||||
|
- влаги |
|
|||||||
|
- общего сахара |
|
|||||||
|
Кислотность |
|
ГОСТ 3624-67 |
||||||
|
Температура |
|
Измерением |
||||||
|
Фосфатаза |
|
|||||||
|
мороженое мягкое |
ОСТ 28 2-77 |
Массовая доля: |
|
|
||||
|
- жира |
|
|||||||
|
- сахарозы |
|
|||||||
|
- сухих веществ |
|
|||||||
|
Кислотность |
|
ГОСТ 3624-67 |
||||||
|
Молочные консервы: |
|
|
|
|
||||
|
молоко цельное сгущенное с сахаром |
Массовая доля: |
|
|
|||||
|
- влаги |
|
|||||||
|
- сахарозы |
||||||||
|
- общего количества сухи веществ, в т.ч. жира |
||||||||
|
- солей свинца, олова, меди |
|
|
||||||
|
Кислотность |
|
|
||||||
|
Вязкость |
|
|||||||
|
Чистота восстановленного сгущенного молока по эталону |
||||||||
|
Дополнительно: размеры кристаллов молочного сахара |
|
|
||||||
|
сливки сгущенные с сахаром |
Массовая доля: |
|
|
|||||
|
- влаги |
|
|||||||
|
- сахарозы |
||||||||
|
- общего количества сухи веществ, в т.ч. жира |
||||||||
|
- солей свинца, олова, меди |
||||||||
|
Кислотность |
|
|||||||
|
Чистота восстановленного сгущенного молока по эталону |
||||||||
|
кофе со сгущенным молоком и сахаром |
То же |
То же |
||||||
|
какао со сгущенным молоком и сахаром |
То же и вязкость |
То же |
||||||
|
молоко цельное сухое |
Массовая доля: |
|
|
|||||
|
- влаги |
|
|||||||
|
- жира |
||||||||
|
- белка |
ГОСТ 23327-78 или 25179-82 |
|||||||
|
Индекс растворимости |
|
|||||||
|
Кислотность |
||||||||
|
Чистота |
||||||||
|
Массовая доля солей олова, меди, свинца |
|
|||||||
|
|
|
|||||||
|
|
|
|||||||
|
Сыры: |
|
|
|
|
||||
|
сыры сычужные твердые |
Массовая доля: |
|
|
|||||
|
- жира |
|
|||||||
|
- влаги |
|
|||||||
|
- поваренной соли |
|
|||||||
|
сыр российский |
То же |
То же |
||||||
|
5. |
РЫБА И РЫБНЫЕ ПРОДУКТЫ: |
|
|
|
|
|||
|
рыба живая |
Длина рыб |
|
ГОСТ 1368-55, |
|||||
|
рыба охлажденная |
То же |
ГОСТ 8636-85, |
||||||
|
рыба мороженая |
Пестициды, гельминты |
|||||||
|
|
|
Длина рыб |
|
ГОСТ 1368-55 |
||||
|
филе рыбное мороженое |
ГОСТ 3948-82 |
Температура в толще филе |
|
|||||
|
наборы рыбные для ухи мороженые |
То же |
|
То же |
|||||
|
рыба океанического промысла мороженая |
То же и массовая доля жира в мясе курильской скумбри |
|
То же |
|||||
|
сардины мороженые |
ГОСТ 21230-75 |
Длина сардин |
|
ГОСТ 1368-55 |
||||
|
|
|
Температура в толще блока |
|
|||||
|
рыба специальной разделки мороженая |
Температура в толще блока |
|
||||||
|
рыба специальной разделки незамороженная |
ОСТ 15 37-72 |
Массовая доля поваренной соли (в случае фиксации) |
|
То же |
||||
|
паста белковая мороженая "Океан" |
Массовая доля воды |
|
||||||
|
Консервы рыбные: |
|
|
|
|
||||
|
консервы рыбные натуральные |
Массовая доля: |
|
|
|||||
|
- поваренной соли |
|
|||||||
|
- солей олова, свинца |
|
|||||||
|
|
|
|||||||
|
Консервы. Креветки натуральные |
ГОСТ 18656-88 |
Массовая доля: |
|
|
||||
|
- поваренной соли |
|
|||||||
|
- мяса |
|
|||||||
|
- солей тяжелых металлов |
|
|||||||
|
|
|
|||||||
|
Определение остаточных количеств пестицидов |
|
|
||||||
|
крабы в собственном соку |
Массовая доля: |
|
|
|||||
|
- составных частей |
|
|||||||
|
- олова, свинца, ртути |
|
|||||||
|
|
|
|||||||
|
|
|
|||||||
|
Остаточное количество пестицидов |
|
|
||||||
|
рыба в желе |
Массовая доля: |
|
|
|||||
|
- поваренной соли |
|
|||||||
|
- солей олова, свинца |
|
|||||||
|
|
|
|||||||
|
Кислотность консервов, в рецептуру которых входит уксусная кислота |
|
|||||||
|
|
|
ГОСТ 8756.15-70 |
||||||
|
шпроты в масле |
Массовая доля: |
|
|
|||||
|
- поваренной соли |
|
|||||||
|
- солей олова, свинца |
|
|||||||
|
|
|
|||||||
|
- отстоя в масле к массе рыбы и отстоя |
|
ГОСТ 20221-74 |
||||||
|
- рыбы, масла |
|
|||||||
|
сардины в масле |
Массовая доля: |
|
|
|||||
|
- поваренной соли |
|
|||||||
|
- отстоя в масле к массе рыбы и отстоя |
|
ГОСТ 20221-74, |
||||||
|
|
|
|||||||
|
консервы рыбные в томатном соусе |
Массовая доля: |
|
|
|||||
|
- сухих веществ |
|
|||||||
|
- поваренной соли |
|
|||||||
|
- составных частей |
|
|||||||
|
Кислотность |
|
|||||||
|
Токсичные элементы |
|
|||||||
|
|
|
|||||||
|
консервы из печени рыб |
Массовая доля: |
|
|
|||||
|
- поваренной соли |
|
ГОСТ 25207-87 |
||||||
|
- солей олова, свинца, меди |
|
|||||||
|
|
|
|||||||
|
|
|
|||||||
|
Кислотность |
|
|||||||
|
печень рыб с растительными добавками |
То же |
|
То же |
|||||
|
Пресервы рыбные: |
|
|
|
|
||||
|
сельдь специального посола |
Массовая доля: |
|
|
|||||
|
- поваренной соли |
|
|||||||
|
- жира в мясе рыбы |
|
|||||||
|
- бензойнокислого натрия |
|
|||||||
|
- составных частей |
|
|||||||
|
Буферность |
|
|||||||
|
Длина сельди |
|
ГОСТ 1368-55 |
||||||
|
Токсичные элементы |
|
|||||||
|
Икра: |
|
|
|
|
||||
|
икра лососевая зернистая баночная |
Массовая доля: |
|
|
|||||
|
- поваренной соли |
|
|||||||
|
- антисептиков (уротропина, сорбиновой кислоты) |
|
|||||||
|
Посторонние примеси |
|
Визуально |
||||||
|
Герметичность |
|
|||||||
|
икра зернистая осетровых рыб баночная |
Массовая доля: |
|
|
|||||
|
- поваренной соли |
|
|||||||
|
- солей олова и свинца |
|
|||||||
|
|
||||||||
|
Посторонние примеси |
|
Визуально |
||||||
|
консервы рыборастительные в масле |
Массовая доля: |
|
|
|||||
|
- поваренной соли |
|
|||||||
|
- составных частей |
|
|||||||
|
- солей тяжелых металлов |
|
|||||||
|
консервы рыборастительные в томатном соусе |
То же и массовая доля сухих веществ |
|
||||||
|
Кислотность |
|
|||||||
|
Рыба копченая и соленая: |
|
|
|
|
||||
|
рыба холодного копчения |
Массовая доля: |
|
|
|||||
|
- поваренной соли в мясе рыбы |
|
|||||||
|
- влаги |
||||||||
|
- жира |
||||||||
|
- гистамина в мясе рыбы |
|
|
||||||
|
сельди холодного копчения |
То же, кроме гистамина |
|
То же |
|||||
|
Массовая доля солей тяжелых металлов |
|
|||||||
|
рыба горячего копчения |
Массовая доля: |
|
|
|||||
|
- поваренной соли в мясе рыбы |
|
|||||||
|
- жира в мясе рыбы |
||||||||
|
сельди горячего копчения |
То же и массовая доля солей тяжелых металлов |
|
||||||
|
|
||||||||
|
сельди соленые |
То же |
|
То же |
|||||
|
6. |
КОНЦЕНТРАТЫ ПИЩЕВЫЕ: |
|
|
|
|
|||
|
первые и вторые обеденные блюда |
Массовая доля: |
|
|
|||||
|
- влаги |
|
|||||||
|
- жира |
|
|||||||
|
- минеральных примесей |
|
|||||||
|
- металлопримесей |
||||||||
|
Посторонние примеси |
||||||||
|
Зараженность амбарными вредителями |
||||||||
|
сладкие блюда |
Массовая доля: |
|
|
|||||
|
- влаги |
|
|||||||
|
- общего сахара |
|
|||||||
|
- металлопримесей |
|
|||||||
|
- минеральных примесей |
||||||||
|
Зараженность амбарными вредителями, плесень и посторонние примеси |
||||||||
|
Общая кислотность в пересчете на лимонную кислоту |
|
|||||||
|
7. |
КРУПА: |
|
|
|
|
|||
|
манная |
Влажность |
|
||||||
|
Зольность |
|
|||||||
|
Крупность |
|
|||||||
|
Металломагнитная примесь |
|
|||||||
|
Зараженность вредителями хлебных запасов |
|
|||||||
|
овсяная |
Массовая доля: |
|
|
|||||
|
- влаги |
|
|||||||
|
- доброкачественного зерна |
|
|||||||
|
- необрушенных зерен |
||||||||
|
- сорной примеси |
||||||||
|
- мучки |
||||||||
|
- металломагнитной примеси |
|
|||||||
|
Зараженность вредителями |
|
|||||||
|
В крупе для детского питания еще массовая доля испорченных ядер, минеральные примеси |
|
|||||||
|
Кислотность |
|
|||||||
|
Токсичные элементы |
|
|||||||
|
|
|
|||||||
|
гречневая |
Массовая доля: |
|
|
|||||
|
- влаги |
|
|||||||
|
- доброкачественность ядра |
|
|||||||
|
- нешелушеных зерен |
||||||||
|
- сорной примеси |
||||||||
|
- мучки |
||||||||
|
- испорченных ядер |
||||||||
|
- металломагнитной примеси |
|
|||||||
|
Развариваемость |
|
|||||||
|
Зараженность вредителями |
|
|||||||
|
рисовая |
ГОСТ 6292-70 |
Массовая доля: |
|
|
||||
|
- влаги |
|
|||||||
|
- доброкачественного ядра |
|
|||||||
|
- нешелушеных зерен |
||||||||
|
- сорной примеси |
||||||||
|
- мучки |
||||||||
|
- испорченных ядер |
||||||||
|
- металломагнитной примеси |
||||||||
|
Развариваемость |
|
|||||||
|
Зараженность вредителями, а для детского питания еще: |
|
|||||||
|
Кислотность |
|
|||||||
|
Токсичные элементы |
|
|||||||
|
|
|
|||||||
|
ячменная |
Массовая доля: |
|
|
|||||
|
- влаги |
|
|||||||
|
- доброкачественного ядра |
|
|||||||
|
- сорной примеси |
||||||||
|
- мучки |
||||||||
|
- металломагнитной примеси |
|
|||||||
|
Зараженность амбарными вредителями |
|
|||||||
|
кукурузная |
То же и массовая доля: |
|
|
|||||
|
- свободного зародыша |
|
|||||||
|
- крупы с остатками оболочек и зародыша |
||||||||
|
- целых необработанных зерен кукурузы |
||||||||
|
Зольность |
|
|||||||
|
фасоль продовольственная |
Массовая доля: |
|
|
|||||
|
- влаги |
|
|||||||
|
- сорной примеси |
|
|||||||
|
- зерновой примеси |
||||||||
|
Зараженность вредителями хлебных запасов |
|
|||||||
|
пшено шлифованное |
Массовая доля: |
|
|
|||||
|
- влаги |
|
|||||||
|
- сорной примеси |
|
|||||||
|
- доброкачественных ядер |
||||||||
|
- испорченных ядер |
||||||||
|
- нешелушенных зерен |
||||||||
|
Зараженность амбарными вредителями |
||||||||
|
Семена гелиотропа |
|
|||||||
|
пшеничная ("Полтавская", "Артек") |
То же, кроме нешелушеных зерен, а также массовая доля: |
|
|
|||||
|
- обработанных зерен ржи и ячменя |
|
|||||||
|
- металломагнитной примеси |
|
|
||||||
|
горох шелушеный (лущеный) |
Массовая доля: |
|
|
|||||
|
- влаги |
|
|||||||
|
- сорной примеси |
|
|||||||
|
- изъеденных семян |
||||||||
|
- нешелушеных семян |
||||||||
|
- дробленого гороха |
||||||||
|
- сечки и мучки |
||||||||
|
- металломагнитной примеси |
|
|||||||
|
Зараженность вредителями хлебных запасов |
|
|||||||
|
толокно овсяное |
Массовая доля: |
|
|
|||||
|
- влаги |
|
|||||||
|
- золы |
|
|||||||
|
- минеральной примеси |
|
|||||||
|
- метталомагнитной примеси |
|
|||||||
|
Крупность помола |
|
|||||||
|
Зараженность вредителями хлебных запасов |
|
|||||||
|
Для детского питания еще: |
|
|
||||||
|
Кислотность |
|
|||||||
|
Массовая доля тяжелых металлов |
|
|||||||
|
|
|
|||||||
|
хлопья овсяные |
ГОСТ 21149-75 |
Массовая доля: |
|
|
||||
|
|
- влаги |
|
||||||
|
- золы |
|
|||||||
|
- сорной примеси |
|
|||||||
|
- метталомагнитной примеси |
|
|||||||
|
Развариваемость |
|
|||||||
|
Кислотность |
|
|||||||
|
Зараженность вредителями хлебных запасов |
|
|||||||
|
8. |
ИЗДЕЛИЯ МАКАРОННЫЕ |
ГОСТ 875-69 |
Влажность |
|
||||
|
Кислотность |
||||||||
|
Прочность макарон |
||||||||
|
Содержание: |
||||||||
|
- лома |
||||||||
|
- деформированных изделий |
||||||||
|
- крошки |
||||||||
|
- металлопримесей |
||||||||
|
Наличие амбарных вредителей |
|
|
||||||
|
9. |
МУКА, КРАХМАЛ: |
|
|
|
|
|||
|
мука пшеничная хлебопекарная |
Массовая доля: |
|
|
|||||
|
- влаги |
|
ГОСТ 9404-86 |
||||||
|
- золы в пересчете на сухое вещество |
|
|||||||
|
- сырой клейковины |
|
|||||||
|
- металломагнитной примеси |
|
|||||||
|
Крупность помола |
|
|||||||
|
Зараженность вредителями хлебных запасов |
|
|||||||
|
крахмал картофельный |
Массовая доля: |
|
|
|||||
|
- влаги |
|
ГОСТ 7698-78 |
||||||
|
- общей золы в пересчете на сухое вещество |
||||||||
|
- сернистого ангидрида |
||||||||
|
Кислотность |
||||||||
|
Примеси других видов крахмала |
||||||||
|
Количество крапин на 1 дм2 поверхности крахмала |
||||||||
|
крахмал кукурузный |
Массовая доля: |
|
|
|||||
|
- влаги |
|
ГОСТ 7698-78 |
||||||
|
- золы |
||||||||
|
- протеина |
||||||||
|
- сернистого ангидрида |
||||||||
|
Кислотность |
||||||||
|
Цветная реакция с йодом |
||||||||
|
10. |
ХЛЕБ, ХЛЕБОБУЛОЧНЫЕ ИЗДЕЛИЯ, ПЕЧЕНЬЕ: |
|
|
|
|
|||
|
|
хлеб из пшеничной муки |
Влажность мякиша |
|
|||||
|
|
|
|
Кислотность мякиша |
|
ГОСТ 5670-51 |
|||
|
|
|
|
Пористость мякиша |
|
ГОСТ 5669-51 |
|||
|
|
|
|
Массовая доля: |
|
|
|||
|
|
|
|
- сахара в пересчете на сухое вещество |
|
||||
|
|
|
|
- жира в пересчете на сухое вещество |
|
||||
|
|
хлеб ржаной, ржано-пшеничный и пшенично-ржаной |
То же, кроме массовой доли жира |
|
То же, кроме ГОСТ 5668-68 |
||||
|
|
изделия булочные |
То же, что и для хлеба из пшеничной муки |
|
То же, кроме ГОСТ 5669-51 |
||||
|
|
изделия хлебобулочные мелкоштучные |
То же, кроме пористости |
|
То же, кроме ГОСТ 5669-51 |
||||
|
|
печенье |
Влажность |
|
|||||
|
|
|
|
Массовая доля: |
|
|
|||
|
|
|
|
- общего сахара в пересчете на сухое вещество |
|
||||
|
|
|
|
- жира в пересчете на сухое вещество |
|
||||
|
|
|
|
- золы |
|
||||
|
|
|
|
- общей сернистой кислоты |
|
||||
|
|
|
|
Щелочность |
|
||||
|
|
|
|
Намокаемость |
|
||||
|
|
изделия кондитерские пряничные |
Влажность |
|
|||||
|
|
|
|
Массовая доля: |
|
|
|||
|
|
|
|
- общего сахара |
|
||||
|
|
|
|
- жира |
|
||||
|
|
|
|
- золы |
|
||||
|
|
|
|
Щелочность |
|
||||
|
|
сухари панировочные |
ОСТ 18-255-75 |
Влажность |
|
||||
|
|
|
|
Кислотность в пересчете сухое вещество |
|
|
|||
|
|
|
|
Крупность помола |
|
||||
|
|
|
|
Содержание металломагнитной примеси |
|
|
|||
|
11. |
САХАР И САХАР-РАФИНАД: |
|
|
|
|
|||
|
|
сахар-песок |
ГОСТ 21-78 |
Массовая доля: |
|
|
|||
|
|
- сахарозы в пересчете на сухое вещество |
|
||||||
|
|
- редуцирующих веществ в пересчете на сухое вещество |
|
||||||
|
|
- золы в пересчете на сухое вещество |
|
ГОСТ 12574-67 |
|||||
|
|
- влаги |
|
||||||
|
|
- ферропримесей |
|
||||||
|
|
Цветность |
|
||||||
|
|
Токсичные элементы |
|
||||||
|
|
|
|
||||||
|
|
сахар-рафинад |
ГОСТ 22-78 |
Массовая доля: |
|
|
|||
|
|
|
|
- сахарозы в пересчете на сухое вещество |
|
||||
|
|
|
|
- редуцирующих веществ в пересчете на сухое вещество |
|
||||
|
|
|
|
- влаги |
|
||||
|
|
|
|
Крепость (временное сопротивление параллепипеда раздробляющему давлению пресса Бонвеча) |
|
||||
|
|
|
|
Полная растворимость в воде при 20 °C кубика 1×1 см |
|||||
|
|
|
|
Токсичные элементы |
|
||||
|
|
|
|
|
|
||||
|
12. |
МАСЛА РАСТИТЕЛЬНЫЕ: |
|
|
|
|
|||
|
масло растительное |
ГОСТ 1129-73 |
Цветное число |
|
ГОСТ 5447-69 |
||||
|
Кислотное число |
|
ГОСТ 5476-74 |
||||||
|
Нежировые примеси (отстой по массе) |
|
ГОСТ 5481-66 |
||||||
|
Фосфоросодержащие вещества |
|
ГОСТ 7824-64 |
||||||
|
Влага и летучие вещества |
|
|||||||
|
Мыло (качественная проба) |
|
|||||||
|
Йодное число |
|
|||||||
|
Неомыляемые вещества |
|
|||||||
|
Температура вспышки экстракционного масла |
|
|||||||
|
масло соевое |
Цветное число |
|
ГОСТ 5447-69 |
|||||
|
Кислотное число |
|
ГОСТ 5476-74 |
||||||
|
Нежировые примеси (отстой по массе) |
|
ГОСТ 5481-66 |
||||||
|
Фосфоросодержащие вещества |
|
ГОСТ 7824-64 |
||||||
|
Влага и летучие вещества |
|
|||||||
|
Мыло (качественная проба) |
|
|||||||
|
Йодное число |
|
|||||||
|
Неомыляемые вещества |
|
|||||||
|
Температура вспышки экстракционного масла |
|
|||||||
|
масло хлопковое рафинированное |
То же |
|
То же |
|||||
|
масло арахисовое |
То же, кроме цветного числа, фосфоросодержащих веществ |
|
То же, кроме ГОСТ 5447-69, ГОСТ 7824-64 |
|||||
|
13. |
ЧАЙ, КОФЕ: |
|
|
|
|
|||
|
чай черный байховый фасованный |
Массовая доля: |
|
|
|||||
|
- влаги |
|
|||||||
|
- водорастворимых экстрактивных веществ |
|
|||||||
|
- металломагнитной примеси |
|
|||||||
|
- тяжелых металлов |
|
|||||||
|
|
|
ГОСТ 26929-86 - 26933-86 |
||||||
|
чай черный байховый нефасованный |
ГОСТ 1937-80 |
То же |
|
То же |
||||
|
кофе натуральный жареный (в зернах и молотый) |
Массовая доля: |
|
|
|||||
|
- влаги |
|
|||||||
|
- общей золы |
|
|||||||
|
- золы, не растворимой в 10 процентной соляной кислоте |
||||||||
|
- экстрактивных веществ |
|
|||||||
|
- кофеина |
||||||||
|
- металлопримесей |
|
|||||||
|
Степень помола, посторонние примеси |
||||||||
|
14. |
ПЛОДООВОЩНЫЕ ПРОДУКТЫ: |
|
|
|
|
|||
|
Овощи свежие: картофель свежий продовольственный |
Размер клубней по наибольшему поперечному диаметру |
|
|
|||||
|
Содержание клубней: |
|
|
||||||
|
- размером менее норм |
|
|
||||||
|
- с израстаниями, наростами, позеленевших |
|
|
||||||
|
- увядших клубней |
|
|||||||
|
- с механическими повреждениями |
||||||||
|
- раздавленных, половинок и частей клубней |
|
|
||||||
|
- поврежденных сельскохозяйственными вредителями |
|
|
||||||
|
- пораженных болезнями |
|
|
||||||
|
- подмороженных, запаренных |
|
|
||||||
|
Наличие: |
|
|
||||||
|
- земли, прилипшей к клубням |
|
|
||||||
|
- органической и минеральной примеси |
|
|
||||||
|
морковь столовая свежая, реализуемая в розничной торговой сети |
Размер корнеплодов: |
|
|
|||||
|
- по наибольшему поперечному диаметру |
|
Измерением |
||||||
|
- по длине |
||||||||
|
Содержание корнеплодов: |
|
|
||||||
|
- с отклонениями от установленных размеров |
|
Взвешиванием |
||||||
|
- поломанных, уродливых по форме |
|
|
||||||
|
- с трещинами |
|
|
||||||
|
- загнивших, увядших, запаренных и подмороженных |
|
|
||||||
|
Наличие земли, прилипшей к корнеплодам |
|
|||||||
|
лук репчатый свежий реализуемый |
Размер луковиц по наибольшему поперечному диаметру |
|
Измерением |
|||||
|
Содержание луковиц: |
|
|
||||||
|
- с недостаточно высушенной шейкой |
|
Взвешиванием |
||||||
|
- оголенных |
|
|
||||||
|
- менее установленных размеров |
|
|
||||||
|
- с механическими повреждениями |
|
|
||||||
|
- проросших |
|
|
||||||
|
- загнивших, запаренных подмороженных, поврежденных нематодой и клещами |
|
|||||||
|
свекла столовая свежая, реализуемая в розничной торговой сети |
Размер корнеплодов по наибольшему поперечному диаметру |
|
Измерением |
|||||
|
Содержание корнеплодов: |
|
|
||||||
|
- с отклонениями от установленных размеров |
|
Взвешиванием |
||||||
|
- с механическими повреждениями |
|
|
||||||
|
- увядших, с признаками морщинистости, загнивших, запаренных и подмороженных |
|
|
||||||
|
Наличие земли, прилипшей к корнеплодам |
|
|||||||
|
капуста белокочанная свежая, реализуемая в розничной торговой сети |
Длина кочерыги над кочаном |
|
Измерением |
|||||
|
Масса зачищенного кочана |
|
|
||||||
|
Содержание кочанов: |
|
|
||||||
|
- с механическими повреждениями на глубину 1 - 2 листьев |
|
|
||||||
|
- с сухим загрязнением, механическими повреждениями на глубину 3 листьев, с засечкой кочана и кочерыги |
|
Взвешиванием |
||||||
|
- с механическими повреждениями глубиной свыше 3 листьев, треснувших, загнивших, запаренных, мороженных |
|
|
||||||
|
картофель хрустящий "Московский" |
ОСТ 18-256-75 |
Влажность |
|
|||||
|
Массовая доля: |
|
|
||||||
|
- жира |
|
|||||||
|
- поваренной соли |
|
|||||||
|
Сушеные ягоды, фрукты, яблоки сушеные |
ГОСТ 7336-91 |
|
|
|
||||
|
фрукты косточковые сушеные |
Массовая доля: |
|
|
|||||
|
- влаги |
|
ГОСТ 8756.2-82 |
||||||
|
- сернистого ангидрида |
|
ГОСТ 25555.5-82 |
||||||
|
- токсичных элементов |
|
|||||||
|
|
|
|||||||
|
Количество плодов в 1 кг |
|
|||||||
|
Посторонние примеси |
|
Визуально |
||||||
|
фрукты семечковые сушеные |
То же |
|
То же |
|||||
|
Маринованные и соленые фрукты, овощи и грибы: |
|
|
|
|
||||
|
капуста квашеная |
Массовая доля: |
|
|
|||||
|
- капусты (после свободного стекания сока) по отношению к общей массе с соком |
|
|||||||
|
- поваренной соли |
|
ГОСТ 12230-66 |
||||||
|
- целых кочанов по отношению к массе измельченной капусты |
|
|
||||||
|
Общая кислотность в пересчете на молочную кислоту |
|
ГОСТ 12229-66 |
||||||
|
Посторонние примеси |
|
Визуально |
||||||
|
огурцы соленые |
Массовая доля: |
|
|
|||||
|
- составных частей |
|
|||||||
|
- поваренной соли |
|
ГОСТ 12230-66 |
||||||
|
Общая кислотность |
|
ГОСТ 12229-66 |
||||||
|
помидоры соленые |
То же |
|
То же |
|||||
|
Консервы. Грибы маринованные и отварные |
Массовая доля: |
|
|
|||||
|
- грибов от массы нетто консервов |
|
|||||||
|
- хлоридов |
|
ГОСТ 26186-04 |
||||||
|
- титруемых кислот |
|
|||||||
|
- примесей растительного происхождения |
|
|||||||
|
- минеральных примесей рН маринованных грибов |
|
|||||||
|
|
|
|||||||
|
Посторонние примеси |
|
Визуально |
||||||
|
Токсичные элементы |
|
|||||||
|
|
|
|||||||
|
яблоки моченые |
ОСТ 10 88-87 |
Массовая доля: |
|
|
||||
|
- яблок, % от общей массы с рассолом |
|
|||||||
|
- хлоридов |
|
|||||||
|
- титруемых кислот в расчете на молочную кислоту |
|
|||||||
|
- этилового спирта |
|
ГОСТ 25555.2-82 |
||||||
|
- летучих кислот в рассоле (в расчете на уксусную кислоту) |
|
|||||||
|
- сахаров |
|
ГОСТ 8756.13-70 (раздел 3) |
||||||
|
- сорбиновой кислоты |
|
|||||||
|
Посторонние примеси |
|
Визуально |
||||||
|
Консервы овощные: |
|
|
|
|
||||
|
горошек зеленый консервированный |
Массовая доля: |
|
|
|||||
|
- горошка от массы нетто консервов |
|
|||||||
|
- хлоридов |
|
|||||||
|
Минеральные примеси |
|
|||||||
|
Посторонние примеси |
||||||||
|
Содержание растительных примесей (лепестки, обрывки створок, стручков) |
|
|||||||
|
икра овощная |
Массовая доля: |
|
|
|||||
|
- сухих веществ |
|
ГОСТ 8756.2-82 |
||||||
|
- жира |
|
|||||||
|
- хлоридов |
|
|||||||
|
- тяжелых металлов (медь, олово, свинец) |
|
|||||||
|
|
|
ГОСТ 26932-86 - 26935-86 |
||||||
|
Титруемая кислотность |
|
|||||||
|
томаты целые очищенные стерилизованные |
ГОСТ 26326-84 |
Размер плодов, масса нетто плодов |
|
|
||||
|
Массовая доля: |
|
|
||||||
|
- поваренной соли |
|
ГОСТ 8756.20-70 |
||||||
|
- сухих веществ |
|
ГОСТ 8756.2-82 |
||||||
|
- тяжелых металлов |
|
|||||||
|
|
||||||||
|
pH заливки |
|
ГОСТ 8756.16-70 |
||||||
|
патиссоны консервированные |
ОСТ 10-112-88 |
Массовая доля: |
|
|
||||
|
- патиссонов от массы нетто консервов |
|
|||||||
|
- пряностей от массы консервов |
||||||||
|
- хлоридов |
|
|||||||
|
- титруемых кислот в пересчете на уксусную кислоту |
|
|||||||
|
- минеральных примесей |
|
|||||||
|
- примесей растительного происхождения |
|
|||||||
|
- тяжелых металлов |
|
|||||||
|
|
|
|||||||
|
свекла и морковь гарнирные |
ОСТ 18-34-71 |
Массовая доля: |
|
|
||||
|
- овощей от массы нетто консервов |
|
|||||||
|
- тяжелых металлов (олово, свинец) |
|
|||||||
|
Посторонние примеси |
|
Визуально |
||||||
|
пюре из шпината, щавеля и смеси шпината и щавеля |
ОСТ 10-78-87 |
Массовая доля: |
|
|
||||
|
- растворимых сухих веществ |
|
ГОСТ 8756.2-82 |
||||||
|
- тяжелых металлов (олово, медь, свинец) |
|
|||||||
|
|
||||||||
|
|
||||||||
|
- минеральных примесей |
|
ГОСТ 25555.3-82 (раздел 2) |
||||||
|
- посторонние примеси растительного происхождения |
|
|||||||
|
pH |
|
ГОСТ 26188-84 по п. 4.4 |
||||||
|
Посторонние примеси (кроме минеральных и растительного происхождения) |
|
ОСТ 10-78-87 визуально |
||||||
|
Томатопродукты: |
|
|
|
|
||||
|
продукты томатные концентрированные |
Массовая доля: |
|
|
|||||
|
- растворимых сухих веществ |
|
ГОСТ 8756.2-82 |
||||||
|
- сахаров |
|
ГОСТ 8756.13-70 |
||||||
|
- хлоридов |
|
|||||||
|
- минеральных примесей |
|
ГОСТ 25555.3-82 (раздел 4) |
||||||
|
- примесей растительного происхождения |
|
|||||||
|
Токсичные элементы |
|
|||||||
|
|
||||||||
|
Титруемая кислотность |
|
|||||||
|
Посторонние примеси |
|
Визуально |
||||||
|
соусы томатные |
Массовая доля: |
|
|
|||||
|
- сухих веществ |
|
ГОСТ 8756.2-82 |
||||||
|
- поваренной соли |
|
ГОСТ 8756.20-70 |
||||||
|
- сорбиновой кислоты |
|
|||||||
|
- жира |
|
|||||||
|
- минеральных примесей |
|
|||||||
|
- тяжелых металлов (меди, свинца, олова) |
|
|||||||
|
|
|
|||||||
|
Титруемая кислотность |
|
|||||||
|
Компоты, соки, консервы, плодовые и ягодные: |
|
|
|
|
||||
|
компоты из плодов, ягод, ревеня и дыни |
ГОСТ 816-81 |
Массовая доля: |
|
|
||||
|
- плодов от массы нетто, указанной на этикетке |
|
|||||||
|
- растворимых сухих веществ в сиропе |
|
ГОСТ 8756.2-82 |
||||||
|
- витамина C (в компоте добавлением аскорбинов кислоты) |
|
ГОСТ 24556-81 |
||||||
|
Токсичные элементы |
|
|||||||
|
|
|
|||||||
|
компот из очищенных персиков без косточек |
ГОСТ 26325-84 |
Размер плода |
|
|
||||
|
Масса нетто плодов, %, к указанной на этикетке массе нетто готового продукта |
|
ГОСТ 816-81 |
||||||
|
Массовая доля: |
|
|
||||||
|
- сухих веществ |
|
|
||||||
|
- тяжелых металлов (меди, олова) |
|
|||||||
|
|
||||||||
|
компоты-ассорти |
ОСТ 18-301-76 |
Соотношение массы смеси плодов, ягод или плодов и ягод к массе нетто готового продукта |
|
|||||
|
Массовая доля: |
|
|
||||||
|
- сухих веществ в сиропе |
|
ГОСТ 8756.2-82 |
||||||
|
- солей тяжелых металлов (меди, олова, свинца) |
|
|||||||
|
|
||||||||
|
|
||||||||
|
сок виноградный натуральный |
Массовая доля: |
|
|
|||||
|
- сухих веществ |
|
ГОСТ 8756.2-82 |
||||||
|
- осадка |
|
|||||||
|
- метавинной кислоты |
|
По п. 3.4 ГОСТ 25892-83 |
||||||
|
- сорбиновой кислоты |
|
|||||||
|
- этилового спирта |
|
ГОСТ 25555.2-82 |
||||||
|
- тяжелых металлов (меди, олова, свинца) |
|
|||||||
|
|
|
|||||||
|
|
|
|||||||
|
Титруемая кислотность |
|
|||||||
|
консервы плодовые и ягодные для детского питания |
Массовая доля: |
|
|
|||||
|
- сухих веществ |
|
ГОСТ 8756.2-82 |
||||||
|
- титруемых кислот в расчете на яблочную кислоту |
|
|||||||
|
- жира |
|
|||||||
|
- витамина C |
|
ГОСТ 24556-81 |
||||||
|
- каротина |
|
|||||||
|
- мякоти (в соках) |
|
|||||||
|
Минеральные примеси |
|
|||||||
|
Примеси растительного происхождения |
|
|||||||
|
Токсичные элементы |
|
|||||||
|
|
|
|||||||
|
соки плодово-ягодные купажированные |
ОСТ 18-12-70 |
Массовая доля: |
|
|
||||
|
- сухих веществ |
|
ГОСТ 8756.2-82 |
||||||
|
- мякоти |
|
|||||||
|
- витамина C |
|
|||||||
|
- спирта |
|
|||||||
|
- сорбиновой кислоты |
|
|||||||
|
Кислотность в пересчете на яблочную кислоту |
|
ГОСТ 8756.15-70 |
||||||
|
Токсичные элементы |
|
|||||||
|
|
|
|||||||
|
Посторонние примеси |
|
Визуально |
||||||
|
Джемы, варенье, повидло: |
|
|
|
|
||||
|
джемы |
ГОСТ 7009-83 |
Массовая доля: |
|
|
||||
|
- растворимых сухих веществ |
|
ГОСТ 8756.2-82 |
||||||
|
- титруемых кислот |
|
|||||||
|
- сорбиновой кислоты |
|
|||||||
|
- сернистого ангидрида |
|
ГОСТ 25555.5-82 |
||||||
|
- минеральных примесей |
|
|||||||
|
- примесей растительного происхождения |
|
|||||||
|
Токсичные элементы |
|
|||||||
|
|
|
|||||||
|
повидло |
То же и массовая доля бензойнокислого натрия |
|
То же и ГОСТ |
|||||
|
Посторонние примеси |
|
Визуально |
||||||
|
варенье |
Массовая доля: |
|
|
|||||
|
- плодов от массы нетто |
|
|||||||
|
- растворимых сухих веществ |
|
ГОСТ 8756.2-82 |
||||||
|
- сернистого ангидрида |
|
ГОСТ 25555.5-82 |
||||||
|
- сорбиновой кислоты |
|
|||||||
|
- минеральных примесей |
|
|||||||
|
- примесей растительного происхождения |
|
|||||||
|
Постороние примеси |
|
Визуально |
||||||
|
Токсичные элементы |
|
|||||||
|
|
|
|||||||
|
Фрукты свежие: апельсины |
Размер плодов |
|
Измерением |
|||||
|
мандарины |
Содержание плодов с отклонениями по качеству |
|
Визуально, взвешиванием |
|||||
|
лимоны |
Остаточное количество пестицидов |
|
По методам Минздрава СССР |
|||||
|
15. |
НАПИТКИ БЕЗАЛКОГОЛЬНЫЕ |
Массовая доля: |
|
|
||||
|
- сухих веществ |
|
|||||||
|
- двуокиси углерода |
|
|||||||
|
- спирта |
|
|||||||
|
Кислотность |
|
|||||||
|
Стойкость |
|
|||||||
|
Полнота налива в бутылки |
|
|||||||
|
Давление двуокиси углерода в бутылке |
|
|||||||
|
Токсичные элементы |
|
|||||||
|
16. |
НАПИТКИ СЛАБОАЛКОГОЛЬНЫЕ, ВИНА, КОНЬЯКИ, ВОДОЧНЫЕ ИЗДЕЛИЯ: |
|
|
|
|
|||
|
пиво |
Массовая доля: |
|
|
|||||
|
- сухих веществ в начальном сусле |
|
|||||||
|
- спирта |
|
|||||||
|
- двуокиси углерода |
|
|||||||
|
Кислотность |
|
|||||||
|
Стойкость пива |
|
|||||||
|
Время дображивания |
|
|||||||
|
вина виноградные |
ГОСТ 7208-84 |
Объемная доля этилового спирта |
|
|||||
|
Массовая концентрация: |
|
|
||||||
|
- сахаров |
|
|||||||
|
- титруемых кислот в пересчете на винную кислоту |
|
|||||||
|
Токсичные элементы |
|
|||||||
|
Относительная плотность |
|
|||||||
|
советское шампанское |
То же и массовая концентрация: |
|
|
|||||
|
- летучих кислот |
|
|||||||
|
- общей сернистой кислоты |
|
|||||||
|
- железа |
|
|||||||
|
|
|
ГОСТ 26928-85 |
||||||
|
Давление двуокиси углерода в бутылке |
|
|||||||
|
коньяки |
ГОСТ 13741-78 |
Крепость |
|
|||||
|
Массовая концентрация: |
|
|
||||||
|
- сахара |
|
|||||||
|
- метанола |
|
|||||||
|
- тяжелых металлов (меди, железа) |
|
|||||||
|
|
||||||||
|
Полнота налива в бутылки |
|
|||||||
|
Относительная плотность |
|
|||||||
|
водки и водки особые |
Крепость, щелочность |
|
ГОСТ 5363-82 |
|||||
|
Массовая концентрация: |
||||||||
|
- альдегидов |
||||||||
|
- сивушного масла |
||||||||
|
- эфиров |
||||||||
|
- метилового спирта |
||||||||
|
17. |
КОНЦЕНТРАТ КРАСНОГО СУСЛА, КОНЦЕНТРАТЫ И ЭКСТРАКТЫ КВАСОВ |
Массовая доля сухих веществ |
|
|||||
|
Кислотность |
|
|
||||||
|
Токсичные элементы |
|
|||||||
|
|
||||||||
|
|
||||||||
|
|
||||||||
|
18. |
МАЙОНЕЗЫ |
ОСТ 10-77-87 |
Массовая доля: |
|
|
|||
|
- жира |
|
ОСТ 10-77-87 |
||||||
|
- влаги |
||||||||
|
Кислотность в пересчете на уксусную и лимонную кислоту |
||||||||
|
Стойкость эмульсии |
|
|
||||||
|
pH |
|
|
||||||
|
19. |
КОНСЕРВЫ ДЛЯ ОБЩЕСТВЕННОГО ПИТАНИЯ: |
|
|
|
|
|||
|
салаты и винегреты |
ОСТ 18-33-70 |
Массовая доля: |
|
|
||||
|
- жира |
|
|||||||
|
- поваренной соли |
|
ГОСТ 8756.20-70 |
||||||
|
- жидкой части по отношению к массе нетто |
|
|||||||
|
- сухих веществ |
|
ГОСТ 8756.2-82 |
||||||
|
- тяжелых металлов (олова, свинца) |
|
|||||||
|
|
||||||||
|
Общая кислотность |
|
ГОСТ 8756.15-70 |
||||||
|
Посторонние примеси |
|
Визуально |
||||||
|
салаты для общественного питания |
ОСТ 10.100-87 |
Массовая доля: |
|
|
||||
|
- основной составной части |
|
|||||||
|
- жидкой части |
||||||||
|
- жира |
||||||||
|
- титруемых кислот |
|
|||||||
|
- хлоридов |
|
|||||||
|
- тяжелых металлов (меди, олова, свинца) |
|
|||||||
|
|
||||||||
|
|
||||||||
|
Посторонние примеси |
|
Визуально |
||||||
|
Масса консервов нетто |
|
Взвешиванием |
||||||
|
20. |
МЕД НАТУРАЛЬНЫЙ |
Массовая доля: |
|
|
||||
|
- воды |
|
|
||||||
|
- редуцирующих сахаров (к безводному веществу) |
|
|||||||
|
- сахарозы (к безводному веществу) |
||||||||
|
- олова |
||||||||
|
Диастазное число (к безводному веществу) |
|
|||||||
|
Оксиметилфурфурол |
||||||||
|
Качественная реакция на оксиметилфурфурол |
||||||||
|
Механические примеси |
|
|
||||||
|
Признаки брожения |
|
|
||||||
|
21. |
ВКУСОВЫЕ, КОНСЕРВИРУЮЩИЕ И СКЛЕИВАЮЩИЕ ВЕЩЕСТВА: |
|
|
|
|
|||
|
кислота лимонная пищевая |
Массовая доля: |
|
|
|||||
|
- лимонной кислоты в пересчете на моногидрат |
|
|||||||
|
- золы |
||||||||
|
- свободной серной кислоты |
||||||||
|
- сульфатной золы |
||||||||
|
Токсичных элементов |
|
|||||||
|
|
|
|||||||
|
Цвет |
|
|
||||||
|
Пробы: на оксалаты, на барий, на ферроцианиды, на сульфаты, на легкообугливаемые вещества, на железо |
|
|
||||||
|
ванилин |
Растворимость: |
|
|
|||||
|
- в воде |
|
|||||||
|
- в спирте |
||||||||
|
- в серной кислоте |
||||||||
|
Температура плавления |
|
|||||||
|
Массовая доля: |
|
|
||||||
|
- ванилина |
|
|||||||
|
- золы |
||||||||
|
соль поваренная пищевая |
ГОСТ 13830-84 |
Массовая доля: |
|
|
||||
|
- хлористого натрия |
|
|
||||||
|
- кальцийиона |
|
|
||||||
|
- магнийиона |
|
|
||||||
|
- сульфатиона |
|
|||||||
|
-калийиона |
||||||||
|
- оксида железа (III) |
||||||||
|
- сульфата натрия |
||||||||
|
- нерастворимых в воде веществ |
|
|
||||||
|
- влаги |
|
|
||||||
|
pH раствора |
|
|
||||||
|
лист лавровый сухой |
Длина листа |
|
|
|||||
|
Влажность листа |
|
|
||||||
|
Массовая доля: |
|
|
||||||
|
- желтых листьев |
|
|||||||
|
- ломаных листьев |
||||||||
|
- листьев с мелкоточечной пятнистостью |
||||||||
|
- минеральной и органической примеси |
|
|
||||||
|
перец черный и белый (молотый и горошком) |
ОСТ 18-279-76 |
Массовая доля: |
|
|
||||
|
- влаги |
|
|
||||||
|
- эфирного масла |
|
|
||||||
|
- золы, нерастворимой в 10 процентной соляной кислоте |
|
|
||||||
|
- мелких и дробленых зерен |
|
для перца горошком |
ГОСТ 18-11-70 |
|||||
|
- плодоножек, оболочек |
|
|
||||||
|
- зерен с плесенью |
|
|
||||||
|
- крупность помола для молотого перца |
|
|
||||||
|
- содержание ферропримесей |
|
|
||||||
|
22. |
ДРОЖЖИ ХЛЕБОПЕКАРНЫЕ ПРЕССОВАННЫЕ |
Влажность |
|
|||||
|
|
|
|
Подъемная сила |
|||||
|
|
|
|
Кислотность в пересчете на уксусную кислоту |
|||||
|
|
|
|
Стойкость |
|||||
_________________
* В связи с тем, что номера стандартов периодически изменяются, их сверяют по Указателям стандартов.
Приложение 4
Перечень общесоюзных санитарно-гигиенических и санитарно-противоэпидемических правил и норм, методических указаний и рекомендаций Минздрава СССР для оценки гигиенических показателей безопасности продовольственного сырья и пищевых продуктов Медико-биологические требования и санитарные нормы качества продовольственного сырья и пищевых продуктов. МЗ СССР, 1990 г.
СанПиН 42-123-4540-87 "Максимально допустимые уровни содержания пестицидов в пищевых продуктах и методы их определения".
СанПиН 42-123-4089-85 "Предельно допустимые концентрации тяжелых металлов и мышьяка в продовольственном сырье и пищевых продуктах".
"Сырье и продукты пищевые. Методы определения токсичных элементов". ГОСТ: 26929-86, 26927-86, 26928-86, 26930-86 - 26935-86.
СанПиН 42-123-4619-88 "Допустимые уровни содержания нитратов в продуктах растительного происхождения и методы их определения".
СанПиН 42-123-4083-86 "Временные гигиенические нормативы содержания гистамина в рыбопродуктах".
"Временные гигиенические нормативы содержания №-нитрозаминов в пищевых продуктах", МЗ СССР, № 4228-85.
"Методические рекомендации по определению химическим методом остаточных количеств диэтилстильбэстрола в продуктах животноводства", МЗ СССР, № 2944-83.
"Методические рекомендации по определению химическим методом остаточных количеств эстрадиола-17 в продуктах животноводства", МЗ СССР, № 3208-85.
"Методические указания по определению остаточных количеств антибиотиков в продуктах животноводства", МЗ СССР, № 3049-84.
"Методические рекомендации по обнаружению, идентификации и определению содержания афлатоксинов в пищевых продуктах", МЗ СССР, № 2273-80.
"Методические указания по обнаружению, идентификации и определению афлатоксинов в продовольственном сырье и пищевых продуктах с помощью высокоэффективной жидкостной хроматографии", МЗ СССР, № 4082-86.
"Методические рекомендации по обнаружению, идентификации и определению содержания патулина в фруктовых и овощных соках и пюре", МЗ СССР, № 2655-82.
"Методические рекомендации по обнаружению, идентификации и определению содержания зеараленона в пищевых продуктах", МЗ СССР, № 2964-84.
"Методические рекомендации по обнаружению, идентификации и определению Т-2 токсина в пищевых продуктах и продовольственном сырье", МЗ СССР, № 3184-84.
"Методические указания по обнаружению, идентификации и определению деоксиниваленона (вомитоксина) в зерне и зернопродуктах", МЗ СССР, № 3940-84.
"Инструкция по проведению ветеринарно-токсикологических, медико-биологических исследований стимуляторов роста сельскохозяйственных животных и гигиенической оценки продуктов животноводства", МЗ СССР, № 3202-85, ГАП СССР № 115-6а.
СОДЕРЖАНИЕ